Psychedelic therapy legalization and clinical access in 2026
The landscape of psychedelic therapy in 2026 represents a critical convergence of clinical research, federal regulatory shifts, and state-level legislative action. Following years of sustained investment and rigorous Phase 3 clinical trials, the medicalization of compounds such as psilocybin, midomafetamine (MDMA), and ibogaine has transitioned from a theoretical frontier to an imminent clinical reality. As of mid-2026, the industry is operating under a dual-track paradigm: a federal pathway focused on stringent, FDA-approved synthetic pharmaceuticals, and a highly fragmented, state-by-state pathway centered on facilitated access to botanical and natural medicines 121.
This report provides an exhaustive analysis of the psychedelic therapy sector as of May 2026. It examines recent federal mandates, late-stage clinical trial efficacy and safety data, the regulatory fallout from highly publicized application rejections, international real-world implementation outcomes, and the structural bottlenecks within clinical infrastructure and insurance reimbursement that will dictate patient access in the coming decade.
Federal Regulatory Framework and Executive Directives
The federal posture toward psychedelic medicine shifted markedly in the spring of 2026, driven by a combination of executive action and targeted regulatory clearances by the U.S. Food and Drug Administration (FDA) and the Drug Enforcement Administration (DEA).
On April 18, 2026, President Donald Trump signed an executive order titled "Accelerating Medical Treatments for Serious Mental Illness" 145. While the order does not legalize psychedelic compounds for broad recreational or clinical use, nor does it bypass the standard evidentiary requirements of the FDA, it mandates several structural changes designed to expedite clinical access, promote data generation, and reduce post-approval regulatory lag 152. The executive order represents a formal acknowledgment of the severe public health burden posed by treatment-resistant psychiatric conditions, particularly among military veterans and first responders, and positions psychedelic medicine as a national research priority 345.
The directive instructs federal agencies to implement four primary mechanisms to accelerate clinical availability. First, the FDA Commissioner was ordered to issue Commissioner's National Priority Vouchers (CNPVs) to psychedelic drug candidates that hold Breakthrough Therapy designation and meet specific public health criteria 5106. These vouchers effectively compress the standard six-to-ten-month regulatory review timeline down to one-to-two months upon submission of a New Drug Application (NDA) 56. Second, the FDA and DEA were directed to establish a formalized pathway for eligible patients to access investigational psychedelic drugs, with explicit mention of ibogaine compounds, under the federal Right to Try Act 527. This provision requires the DEA to facilitate necessary Schedule I handling authorizations for treating physicians 52.
Third, to eliminate the historical delay between FDA approval and commercial availability, the DEA is now required to initiate controlled substance rescheduling reviews in parallel with the FDA review process, triggering immediately upon the successful completion of Phase 3 clinical trials 4210. Finally, the order allocates $50 million through the Advanced Research Projects Agency for Health (ARPA-H) to match state-level investments in psychedelic research, signaling a deliberate effort to leverage state infrastructure for federal data collection 4107.
Professional medical organizations have responded to the federal shift with cautious optimism. The American Psychiatric Association (APA) publicly supported the federal investment but reiterated the necessity of an "evidence-first approach," cautioning that psychedelic administration must remain strictly within controlled clinical settings governed by trained clinicians and subject to rigorous scientific evaluation 8.
Clinical Trial Pipeline and Food and Drug Administration Pathways
Following the executive order, the FDA announced on April 24, 2026, that it had issued CNPVs to three specific advanced programs and authorized new early-phase clinical studies 5915. The current late-stage pipeline is dominated by synthetic analogues of classic psychedelics and entactogens targeting severe psychiatric indications.
| Sponsor | Compound | Target Indication | Clinical Phase | FDA Designations | Target NDA Timeline |
|---|---|---|---|---|---|
| Compass Pathways | COMP360 (Synthetic Psilocybin) | Treatment-Resistant Depression (TRD) | Phase 3 (COMP005/COMP006) | Breakthrough Therapy, CNPV | Q4 2026 Submission |
| Usona Institute | PSIL201/PSIL301 (Synthetic Psilocybin) | Major Depressive Disorder (MDD) | Phase 3 (uAspire) Completed | Breakthrough Therapy, CNPV | Post-2026 Analysis |
| Otsuka / Transcend | TSND-201 (Methylone) | Post-Traumatic Stress Disorder (PTSD) | Phase 3 Enrolling | Breakthrough Therapy, CNPV | Undisclosed |
| Reunion Neuroscience | Luvesilocin (RE104) | Postpartum Depression | Phase 2 Completed | Breakthrough Therapy | Phase 3 in late 2026 |
| Atai / Beckley Psytech | BPL-003 (5-MeO-DMT Nasal Spray) | Treatment-Resistant Depression (TRD) | Phase 2b Completed | Fast Track | Phase 3 in Q2 2026 |
Compass Pathways remains the most advanced sponsor in the psychedelic pharmaceutical space. The company's proprietary synthetic psilocybin formulation, COMP360, has completed two pivotal Phase 3 trials 1011. The initial COMP005 trial, reporting in June 2025, demonstrated that 25% of participants in the 25mg cohort achieved a clinically meaningful response (defined as a ≥25% reduction in Montgomery-Åsberg Depression Rating Scale [MADRS] scores) that remained durable through 26 weeks following one or two administrations 1012. The subsequent global COMP006 trial, which released data in February 2026, confirmed a highly statistically significant 3.8-point reduction in MADRS scores at week six compared to a 1mg control dose (p<0.001) 121314. In this later trial, 39% of participants achieved a clinically meaningful response, with onset of symptom relief occurring as early as the day following administration 1213. The treatment demonstrated a robust safety profile, with severe treatment-emergent adverse events (TEAEs) - primarily transient headaches, nausea, and anxiety - occurring in only 2% to 5% of patients 12. With regulatory alignment on a rolling NDA, Compass anticipates completing its submission in the fourth quarter of 2026 1510.
Operating as a nonprofit drug developer, Usona Institute concluded enrollment for its Phase 3 uAspire trial (PSIL301) in November 2025 15. This trial evaluates single-dose synthetic psilocybin (25mg) combined with psychosocial support for MDD across 30 active clinical sites, importantly including four Veterans Affairs medical centers to broaden geographic reach and participant diversity 1115. Usona's strategy focuses on a broader indication than TRD, aiming to position psilocybin earlier in the psychiatric treatment paradigm 11. With a CNPV granted by the FDA, Usona is mobilizing toward an NDA submission once long-term durability data from the uAspire trial is finalized 915.
The FDA also awarded a CNPV to Otsuka Pharmaceutical and Transcend Therapeutics for TSND-201, an investigational rapid-acting neuroplastogen (methylone) currently enrolling Phase 3 trials for PTSD 91516. Transcend emphasizes that TSND-201 does not act primarily at 5-HT2A receptors and is being developed as a non-hallucinogenic treatment, theoretically bypassing the intensive clinical monitoring requirements that constrain classic psychedelics 416. Additionally, the FDA cleared DemeRx NB to initiate Phase 1 clinical studies of noribogaine hydrochloride - an ibogaine derivative - for alcohol use disorder, marking the first time a clinical study of an ibogaine-derived compound has been permitted on U.S. soil 56.
Regulatory Setbacks and Evolving Clinical Standards
The accelerated pace of the pipeline in 2026 is structurally informed by the regulatory failure of Lykos Therapeutics (formerly MAPS Public Benefit Corporation) in 2024. The FDA's handling of the Lykos application established the contemporary standard for psychedelic drug reviews, fundamentally altering how sponsors design clinical trials.
Despite publishing landmark Phase 3 trials (MAPP1 and MAPP2) demonstrating that over 71% of participants with severe, chronic PTSD no longer met diagnostic criteria following MDMA-assisted therapy, the FDA issued a Complete Response Letter (CRL) rejecting the application in August 2024 171825. The rejection aligned with a 10-1 vote against approval by the FDA's Psychopharmacologic Drugs Advisory Committee 1718. The FDA's CRL, which was subsequently made public in September 2025, highlighted several profound methodological and safety concerns that now serve as the regulatory baseline for the industry 192021.
The primary barrier identified by the FDA was functional unblinding. Due to the profound psychoactive and somatic effects of MDMA, the overwhelming majority of participants and site investigators could accurately guess whether the active drug or a placebo was administered 182223. The agency argued this unblinding, combined with high patient expectations, hopelessly compromised the integrity of subjective patient-reported outcome measures 2223. Furthermore, the FDA identified severe selection bias, noting high rates of prior illicit MDMA use among study participants, suggesting the cohort was predisposed to positive outcomes and potentially masking adverse reactions 2021.
Crucially, the FDA criticized the sponsor's handling of adverse event reporting. Trial safety manuals had instructed investigators to exclude "positive or favorable effects" (such as euphoria or enhanced interpersonal connection) from adverse event logs 19. The FDA deemed these effects essential for assessing the drug's abuse liability under the Controlled Substances Act, characterizing the omission as a fundamental failure to characterize the safety profile of a scheduled substance 1920. The regulatory crisis was exacerbated on August 10, 2024, when the journal Psychopharmacology retracted three papers related to MDMA-assisted therapy due to protocol violations and ethical misconduct at specific study sites, later revealed to involve data handling irregularities by site personnel 2324.
The rejection forced a 75% workforce reduction at Lykos, the departure of founder Rick Doblin, and a corporate restructuring 2325. As of 2025, the company is charting a path forward involving an independent third-party audit of prior data and the initiation of a new Phase 3 trial to address durability and safety characterization 2025. Consequently, the industry recognizes that the FDA views psychedelic therapies through an acutely conservative lens. The agency is currently finalizing guidance on clinical trial design for serotonin-2A agonists, which will formally codify enhanced requirements for managing expectation bias, designing robust control arms, and monitoring cardiac safety during administration 51626.
State-Level Legalization and Implementation Models
While federal authorities navigate the pharmaceutical approval of isolated synthetic molecules, a parallel movement has taken root at the state level. Over 28 states introduced legislation regarding psychedelic access leading into 2026, reflecting a divergence between the slow pace of federal clinical trials and the urgent public demand for novel mental health interventions 27. State approaches range from broad decriminalization and adult-use service models to strict medicalized frameworks requiring physician oversight.
Decriminalization and Supported Adult-Use Models
Oregon was the first state to implement a regulated psychedelic system following the passage of Measure 109. The state operates a service-center model where adults aged 21 and older can access psilocybin for therapeutic or personal development purposes under the supervision of state-licensed facilitators 282930. The Oregon model is strictly non-medical; no clinical diagnosis or physician prescription is required for access 29. By early 2026, over 10,000 clients had utilized the program across roughly 34 licensed service centers 29. However, the model has faced significant economic friction. High regulatory compliance, facilitator licensing fees, and unique liability insurance costs mean that a single session typically ranges from $1,500 to over $3,400 229. As a result, the average client demographic heavily skews toward affluent individuals earning over $160,000 annually, and roughly a third of the initially licensed service centers have closed due to operating deficits and insufficient local demand 129.
Colorado adopted a more expansive framework under the Natural Medicine Health Act (Proposition 122), combining the decriminalization of personal cultivation and sharing with a regulated commercial framework 22838. Operating 34 licensed "healing centers" as of mid-2026, Colorado utilizes a phased approach 229. Psilocybin and psilocin are currently available, with the state preparing for the potential inclusion of DMT, ibogaine, and non-peyote mescaline after June 1, 2026 229. This multi-substance approach positions Colorado as the most comprehensive psychedelic market in the United States. Following legislative clarifications (Senate Bill 290), Colorado also permits professionals to provide paid harm-reduction and integration services alongside the "gifting" of natural medicines, a unique structural advantage over Oregon 28.
Medicalized Frameworks and Equity Initiatives
Diverging from the ballot-initiative adult-use models, New Mexico became the first state to establish psychedelic access through traditional legislative channels by passing the Medical Psilocybin Act (SB 219) in April 2025 23931. New Mexico's model is strictly medicalized. It requires patients to have a qualifying condition - explicitly defined as major treatment-resistant depression, PTSD, substance use disorders, or end-of-life psychological distress - and necessitates a physician's referral and medical supervision 293932.
To address the economic disparities observed in Oregon's free-market approach, the New Mexico legislature established a pioneering Psilocybin Treatment Equity Fund, capitalizing it with an initial $630,000 allocation in March 2026 3233. This fund is designed to subsidize treatment for low-income patients who would otherwise be priced out of care, as federal prohibition prevents Medicaid or commercial insurance from covering the interventions 3233. The state is also heavily focused on incorporating traditional and Indigenous knowledge, requiring the medical advisory board to include representatives from Indian nations and tribes 3931. The New Mexico Department of Health is currently drafting regulations with the goal of enrolling the first patients by December 2026 293234.
Legislative Pilot Programs and Clinical Research
In states hesitant to launch full commercial or medical markets, structured clinical pilot programs have emerged as a dominant legislative strategy. In Massachusetts, while advocacy groups push a broad Colorado-style ballot initiative for the 2026 cycle, state lawmakers have concurrently advanced research-focused pilots 2844. As of March 2026, the legislature advanced H.4200 and H.2203, which authorize the Department of Public Health to issue permits to up to three licensed mental health clinics to administer psychedelics 4535. These clinics must utilize multidisciplinary care teams, partner with university researchers, and rigorously track patient outcomes related to depression, PTSD, and substance use to refine best clinical protocols 4535. Importantly, the legislation stipulates that participating clinics cannot be affiliated with cannabis operations or pharmaceutical developers, ensuring the pilot remains focused purely on clinical outcomes rather than commercialization 4535.
Similar incremental approaches are advancing nationwide. Utah passed HB 390, establishing a donation-funded clinical study on psilocybin-assisted therapy specifically for veterans with TRD and PTSD 44. New Jersey enacted the Psilocybin Behavioral Health Access and Therapy Pilot Program (S2283) with $6 million in hospital-based funding 244. Washington state is reviewing the REACH WA bill to establish safe access pathways, while voters in Alaska will weigh a Natural Medicine Act ballot initiative in late 2026 244.
| State | Legal Mechanism | Access Model | Target Population | Cost/Equity Interventions | Operational Status (May 2026) |
|---|---|---|---|---|---|
| Oregon | Ballot Measure 109 | Supported Adult-Use (Service Centers) | Adults 21+ (No diagnosis required) | None mandated; market-driven high costs ($1.5k - $3.5k) | Fully operational; 10,000+ served. |
| Colorado | Ballot Prop 122 | Decriminalization + Healing Centers | Adults 21+ (No diagnosis required) | Sliding scales offered voluntarily by some centers | Psilocybin active; DMT/Ibogaine pending June 2026. |
| New Mexico | Legislation (SB 219) | Strict Medical Model | Diagnosed patients (TRD, PTSD, SUD, End-of-Life) | State-funded $630k Psilocybin Treatment Equity Fund | Rulemaking phase; targeting Dec 2026 launch. |
| Massachusetts | Pending Legislation (H.4200) | Clinical Pilot Program | Diagnosed patients via 3 approved mental health clinics | Mandated university partnership and data tracking | Bills advanced from committee March 2026. |
| Utah | Legislation (HB 390) | Clinical Research Pilot | Veterans with treatment-resistant PTSD | Donation-funded operational model | Enacted; trial design underway. |
International Paradigms and Real-World Data
The U.S. landscape is heavily informed by early real-world data emerging from international jurisdictions that have cautiously integrated psychedelics into formal medical frameworks. These global experiments offer critical insights into the logistical challenges of widespread implementation.
Australian Down-Scheduling Outcomes
In a landmark global decision, Australia's Therapeutic Goods Administration (TGA) down-scheduled MDMA and psilocybin from prohibited substances to controlled medicines effective July 2023 363749. This permitted specifically authorized psychiatrists to prescribe them for PTSD and TRD outside of clinical trials 364938.
However, the rollout has been characterized by extreme caution, high costs, and significant structural bottlenecks. Through September 2025, freedom of information requests revealed that only 134 unique patients had accessed the treatment (87 for MDMA, 47 for psilocybin) via roughly 13 active Authorised Prescribers nationwide 3637. The deliberate pace is attributed to a highly complex, multi-tiered regulatory process requiring approval from both the TGA and an institutional Human Research Ethics Committee (HREC) 37. Compounding these challenges is the absence of locally registered psilocybin or MDMA products on the Australian Register of Therapeutic Goods (ARTG). Clinics must rely on imported formulations accessed under Section 19A exemptions, creating supply chain vulnerabilities and raising treatment costs well beyond the reach of average citizens 137. New Therapeutic Goods Orders (TGOs) implemented in 2025 mandate strict Good Manufacturing Practice (GMP) compliance and purity limits for imports, further constraining supply 37.
Crucially, however, the clinical safety data emerging from this cohort has been highly encouraging. Through late 2025, zero serious adverse events (SAEs) were reported among the treated cohort, validating the physical safety of the clinical protocols when administered by rigorously trained professionals in controlled medical settings 36. This data suggests that while operationalizing broad access is administratively difficult, the physiological risks of the compounds remain low under strict supervision.
Canada's Special Access Program Constraints
Canada operates a Special Access Program (SAP) which allows healthcare practitioners to request restricted drugs, including psilocybin and MDMA, for patients with serious or life-threatening conditions where conventional therapies have failed or are unavailable 394041. The SAP explicitly recognizes end-of-life distress and TRD as eligible indications for psilocybin, and PTSD for MDMA 3955.
While conceptually more robust than the FDA's expanded access pathway, the Canadian SAP remains highly restrictive in practice. Between the inclusion of psychedelics in 2022 and mid-2025, Health Canada had authorized just 301 requests for psilocybin 56. The severe bureaucratic burden of the application process has led to a widening gap between patient demand and legal access, inadvertently fueling a robust underground network of unregulated practitioners serving veterans and complex psychiatric cases 3. Patient advocacy has led to successful legal challenges against Health Canada; notably, in 2024 and 2025, courts forced the agency to reconsider exemptions for cluster headache patients and for healthcare workers seeking experiential training with the substances 56. Canadian stakeholders point to the U.S. executive order as a potential catalyst that could politically pressure Health Canada to streamline its regulatory apparatus and broaden access parameters 3.
Similarly, European nations have relied on limited special access programs. In Switzerland, hundreds of patients have been treated under limited-use exemptions over the past decade 42. However, broader market approval for these treatments by the European Medicines Agency (EMA) is not anticipated until 2028 or later, leaving Europe reliant on fragmented compassionate use frameworks 42.
Clinical Infrastructure and Workforce Development
Regardless of whether a psychedelic compound achieves FDA approval or state-level authorization, the ultimate constraint on patient access in 2026 is the physical and human infrastructure required to deliver care. Unlike traditional psychiatric medications dispensed at a pharmacy for daily at-home use, psychedelic therapy is a complex, labor-intensive bundle of medical and psychological services 143.
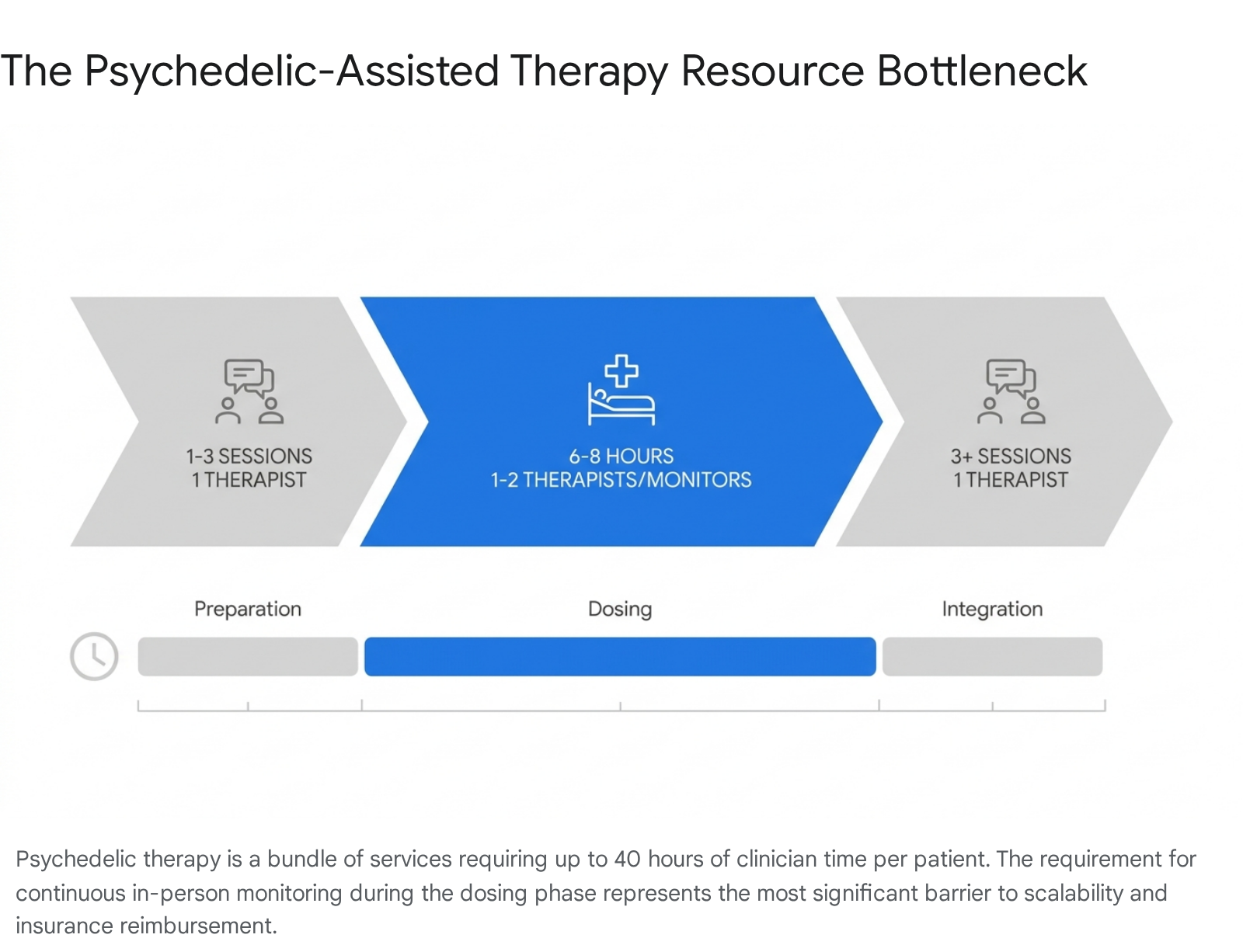
The standard protocol utilized in clinical trials and mandated by emerging regulations involves a dyad model - typically two therapists present for the duration of a six-to-eight-hour dosing session, flanked by multiple preparation and integration therapy sessions 1459. This model requires upwards of 40 hours of highly specialized clinician time per patient 159. Projections indicate that delivering care at scale across the United States will require training over 50,000 specialized practitioners globally over the next decade 59.
Currently, there is no centralized governmental accreditation standard for psychedelic therapists 3744. Instead, the market relies on comprehensive certification programs from independent entities such as Fluence, the California Institute of Integral Studies (CIIS), and MAPS 45944. These programs require extensive training in non-ordinary state facilitation, neuropharmacology, harm reduction, and somatic integration therapy, with leading providers educating thousands of clinicians annually 4446162.
Because the labor costs are exceptionally high - health economists estimate the full MDMA-assisted therapy protocol for PTSD to cost between $12,000 and $48,000 per patient under the dyad model - the industry must inevitably pivot toward scalable delivery methods to make broad insurance coverage viable 1. Implementation scientists and clinical operators suggest that group therapy models are the most likely solution 145. Emerging research indicates that group protocols can reduce per-patient clinician costs by 34% for psilocybin and 51% for MDMA, while requiring significantly fewer full-time equivalent clinicians 1. However, shifting to group models will require the FDA to accept novel efficacy datasets generated outside the individual-dyad framework utilized in pivotal Phase 3 trials 1.
Health Economics and Insurance Reimbursement
Without a viable pathway for commercial insurance or Medicare/Medicaid reimbursement, psychedelic therapy risks remaining an exclusive, out-of-pocket intervention for affluent populations 13746. The American Medical Association (AMA) took a crucial first step in 2024 by activating Category III Current Procedural Terminology (CPT) codes (0820T, 0821T, 0822T, and 0823T) specifically designed to capture the unique workflows of "Continuous In-Person Monitoring and Intervention During Psychedelic Medication Therapy" 43464748.
These Category III codes allow providers to bill for the time of the primary qualified health care professional, as well as modifier codes for additional clinical staff required for the extended dosing observation 4748. Currently, these are temporary tracking codes, meaning they are not yet widely reimbursable under major payer networks. Instead, they are utilized to gather actuarial data on cost, utilization, and clinical outcomes for the Centers for Medicare & Medicaid Services (CMS) 43. Once the FDA officially approves a compound like COMP360, these codes are expected to transition to permanent Category I codes, establishing the relative value units (RVUs) that dictate reliable reimbursement rates 4348. Until that transition occurs, clinics must rely on hybrid billing models - attempting to bill standard evaluation and management (E&M) codes for the psychotherapy components while charging cash for the extended monitoring - which exposes providers to compliance risks and places immense financial burden on the patient 44346.
The immediate delivery infrastructure for future FDA-approved psychedelics will likely be the existing network of interventional psychiatry and ketamine clinics. As of 2026, an estimated 1,473 specialized clinics operate across the United States, primarily treating unipolar depression with off-label intravenous ketamine or FDA-approved intranasal esketamine (Spravato) 495051. These clinics have already operationalized the difficult logistics of extended patient monitoring, federal Risk Evaluation and Mitigation Strategy (REMS) compliance, and specialized aesthetic environments 4. Market analyses indicate that while these clinics represent a $1.6 billion industry, many suffer from operational inefficiencies, leaving significant revenue uncaptured due to underutilized neurocognitive testing codes and poor patient retention modeling 495051. Nevertheless, they provide a vital, pre-existing physical footprint. Industry consensus suggests these facilities are actively preparing to transition into multi-modality centers offering psilocybin and MDMA once legal and billing pathways clear 4.
The future of psychedelic reimbursement will likely pivot toward value-based payment models. Traditional fee-for-service frameworks are fundamentally ill-suited for treatments requiring up to 24 hours of clinical time per patient over a short duration 4. As state Medicaid programs begin exploring ways to separate drug costs from therapy costs, and as federal agencies leverage ARPA-H funding to pilot innovative care models, the financial viability of psychedelics will depend less on the cost of the synthesized molecule and entirely on the cost-effectiveness of the surrounding clinical labor 14.