Will the FDA Approve Moderna's mRNA Flu Vaccine in 2026
The Food and Drug Administration is highly likely to approve Moderna's standalone mRNA flu vaccine, known clinically as mRNA-1010 and expected to be marketed as mFLUSIVA, by its target decision date of August 5, 2026. Following a high-profile regulatory dispute over clinical trial design that briefly derailed the application in early 2026, the agency and the manufacturer reached a bifurcated approval compromise that puts the shot on track for the upcoming fall respiratory season. A crucial public advisory committee meeting scheduled for June 18 will serve as the final public test of the vaccine's safety and efficacy profile before the agency issues its final ruling.
The Global Burden of Influenza and the Limitations of Current Vaccines
Influenza remains one of the most pervasive and resilient respiratory pathogens on the planet, representing a significant and persistent global public health concern. Every year, seasonal influenza causes an estimated 3 to 5 million severe cases of illness globally, resulting in up to 650,000 deaths 11. The burden on the United States healthcare system is particularly immense. During the recent 2024 - 2025 flu season, influenza-related hospitalizations and outpatient visits reached a 15-year high, with more than 545,000 Americans hospitalized due to flu-related complications 2.
Despite widespread public awareness and annual vaccination campaigns, the tools used to fight the virus have remained technologically stagnant for decades. Current influenza vaccines are primarily manufactured using an antiquated process that involves growing the virus inside millions of fertilized chicken eggs - a technique first pioneered in the 1940s 34.
This traditional manufacturing timeline is incredibly slow and resource-intensive. The process takes approximately six months from start to finish 345. Because of this lengthy lead time, the World Health Organization (WHO) and affiliated global health bodies must convene in February or March to predict which viral strains will be circulating during the Northern Hemisphere's autumn and winter seasons 45.
This creates a massive biological vulnerability. Influenza viruses are infamous for their rapid mutation rates, driven by antigenic drift (gradual mutations over time) and antigenic shift (the sudden genetic reassortment between different strains) 1. By forcing human influenza viruses to replicate inside an avian egg environment for months, the virus naturally adapts to its host. This phenomenon, known as "egg adaptation," causes the virus to mutate further 47. Consequently, the virus harvested, deactivated, and placed into a vaccine vial often bears structural differences from the wild-type virus actively spreading from person to person.
This fundamental mismatch is the primary reason why the real-world effectiveness of seasonal flu shots has fluctuated so wildly over the last 15 years, frequently hovering between a meager 19% and 60% efficacy rate 4678.
How Messenger RNA Rewrites the Flu Vaccine Playbook
The introduction of messenger RNA (mRNA) technology to the seasonal influenza landscape promises to fundamentally alter this paradigm. Leveraging the exact technological platform that achieved global success during the COVID-19 pandemic, mRNA vaccines bypass the need to cultivate live viruses altogether.
The Mechanism of Action
Instead of injecting a weakened or inactivated virus into the body to trigger an immune response, mRNA vaccines deliver specific genetic instructions. The messenger RNA is transcribed from a plasmid DNA template and engineered with optimized sequences, including a poly-A tail and a clean CAP1 structure added during in vitro transcription 11. To prevent the body's immune system from prematurely attacking the mRNA as a foreign pathogen, manufacturers replace standard uridines with modified nucleosides (like pseudo-uridine) and subject the RNA to stringent cellulose purification to remove double-stranded RNA contaminants 11.
Once purified, these genetic instructions are encased within lipid nanoparticles (LNPs) - microscopic fat bubbles that protect the fragile mRNA and facilitate its entry into human cells 611. Once inside the cell, the mRNA instructs the ribosomes to manufacture specific viral proteins. For influenza vaccines like Moderna's mRNA-1010, these instructions code for the hemagglutinin (HA) surface glycoproteins of the specific influenza strains recommended by the WHO 189. The immune system recognizes these newly minted, harmless HA proteins as foreign invaders and mounts a robust defense, producing neutralizing antibodies, activating CD4+ Th1 T-cells, and establishing immunological memory 1011.
The Advantage of Speed
The most profound advantage of the mRNA platform is manufacturing speed. Because the production relies on rapid genetic sequencing and biochemical synthesis rather than biological cultivation, the manufacturing timeline is condensed from the traditional six months to just two or three months 512.
This accelerated timeline theoretically allows public health officials and manufacturers to delay the selection of the target strains until much closer to the start of the flu season 512. By reducing the lead time, the risk of before-season antigenic drift is severely minimized, and the risk of egg-adaptive mutations is eliminated entirely 4513.
The Clinical Evidence: Does mFLUSIVA Actually Work Better?
To prove that mFLUSIVA (mRNA-1010) could outperform traditional egg-based and cell-based shots, Moderna launched an extensive, multi-year clinical trial program encompassing tens of thousands of participants across various demographics and flu seasons.
Early Trials and Strain Refinement
The initial path for mRNA-1010 was not without friction. Early Phase 1 and Phase 2 trials demonstrated that while the mRNA platform was highly effective at eliciting immune responses against influenza A strains (such as H1N1 and H3N2), it struggled to generate a sufficiently robust response against influenza B strains 17. First-generation mRNA flu vaccines across the industry encountered this exact hurdle; the B strains require a different immunological threshold to achieve optimal seroconversion 114.
In response, Moderna utilized the agility of the mRNA platform to rapidly update and optimize the vaccine's lipid and antigen formulation to improve its immunogenicity against B/Victoria and B/Yamagata lineages 78.
The Pivotal Phase 3 "Fluent" Study (P304)
The definitive proof of concept arrived with the P304 efficacy study, also known as the Fluent trial. This randomized, double-blind, active-controlled Phase 3 study enrolled 40,805 participants aged 50 and older across 301 clinical sites in 11 countries during the 2024 - 2025 Northern Hemisphere influenza season 91819.
Participants were randomly assigned in a 1:1 ratio to receive either a single dose of Moderna's trivalent mRNA-1010 vaccine or a licensed, standard-dose comparator vaccine (such as Fluarix or Influsplit, depending on regional availability) 89.
The primary endpoint of the study was relative vaccine efficacy (rVE) against influenza-like illness that was subsequently confirmed by an RT-PCR laboratory test within seven days of symptom onset 920. The results, published in the peer-reviewed New England Journal of Medicine in early May 2026, confirmed that mRNA-1010 achieved its primary endpoints and demonstrated statistical superiority over standard-dose vaccines 221.
| Influenza Strain | Relative Vaccine Efficacy (rVE) vs. Standard Dose |
|---|---|
| Overall Trial Population | 26.6% |
| Adults 65 Years and Older | 27.4% |
| Influenza A/H1N1 | 29.6% |
| Influenza A/H3N2 | 22.2% |
| Influenza B/Victoria | 29.1% |
Data sourced from Moderna's Phase 3 Fluent (P304) trial results, analyzing RT-PCR-confirmed illness across specific viral lineages 21821. Note: The B/Yamagata lineage was excluded from the efficacy breakdown as it is no longer included in the seasonal formulation following WHO guidance citing the strain's disappearance from global circulation 1523.
Understanding Relative Vaccine Efficacy (rVE)
While a 26.6% relative vaccine efficacy sounds impressive, it requires precise contextualization to avoid misinterpretation. The trial did not show that the vaccine was only 26.6% effective overall; rather, it showed that it was 26.6% better than the standard-of-care baseline already provided by existing vaccines.
In absolute numbers, more than 97% of the participants in both the mRNA and the standard-dose groups did not develop a lab-confirmed case of the flu during the study period 1924. Specifically, RT-PCR-confirmed influenza occurred in 2.8% of the participants who received the standard-dose flu shot (557 out of 20,124 people). In the mRNA-1010 group, that infection rate dropped to 2.0% (411 out of 20,179 people) 91916.
This represents a 0.8-percentage-point difference in absolute risk reduction 19. However, in epidemiology and public health, moving the needle by 0.8% across a global population of hundreds of millions of people translates to massive reductions in total clinical caseloads, hospital overcrowding, and mortality rates, satisfying the rigorous non-inferiority and superiority criteria established by regulators 21626.
Immunological Depth and Maturation
Beyond sheer efficacy numbers, advanced systems serology profiling indicated that mRNA-1010 drives a qualitatively different immune response than traditional vaccines. Studies comparing mRNA-1010 against FLUAD (a licensed, adjuvanted flu vaccine specifically designed to boost immune response in seniors) revealed that while overall antigen-specific antibody titers were comparable, the mRNA vaccine drove a more rapid "anamnestic humoral maturation" 27.
This means the mRNA vaccine triggered a faster and more refined maturation of B-cells, resulting in a broader array of highly functional antibodies capable of binding to diverse HA variants 27. While FLUAD induced stronger IgG1 responses against influenza B HAs, mRNA-1010 elicited significantly higher IgG1 responses and superior Fc receptor binding profiles to the dominant A/Darwin H3 HA strains 27.
The Reactogenicity Trade-Off: Safety and Side Effects
The primary challenge surrounding the adoption of mRNA influenza vaccines is their reactogenicity profile. Much like the mRNA COVID-19 vaccines, mRNA-1010 triggers a vigorous and immediately noticeable inflammatory response as the immune system encounters the lipid nanoparticles and begins rapidly synthesizing proteins 312.
In the Phase 3 trials, participants receiving the mRNA vaccine reported systemic and local side effects at rates significantly higher than those who received standard inactivated vaccines 192426.
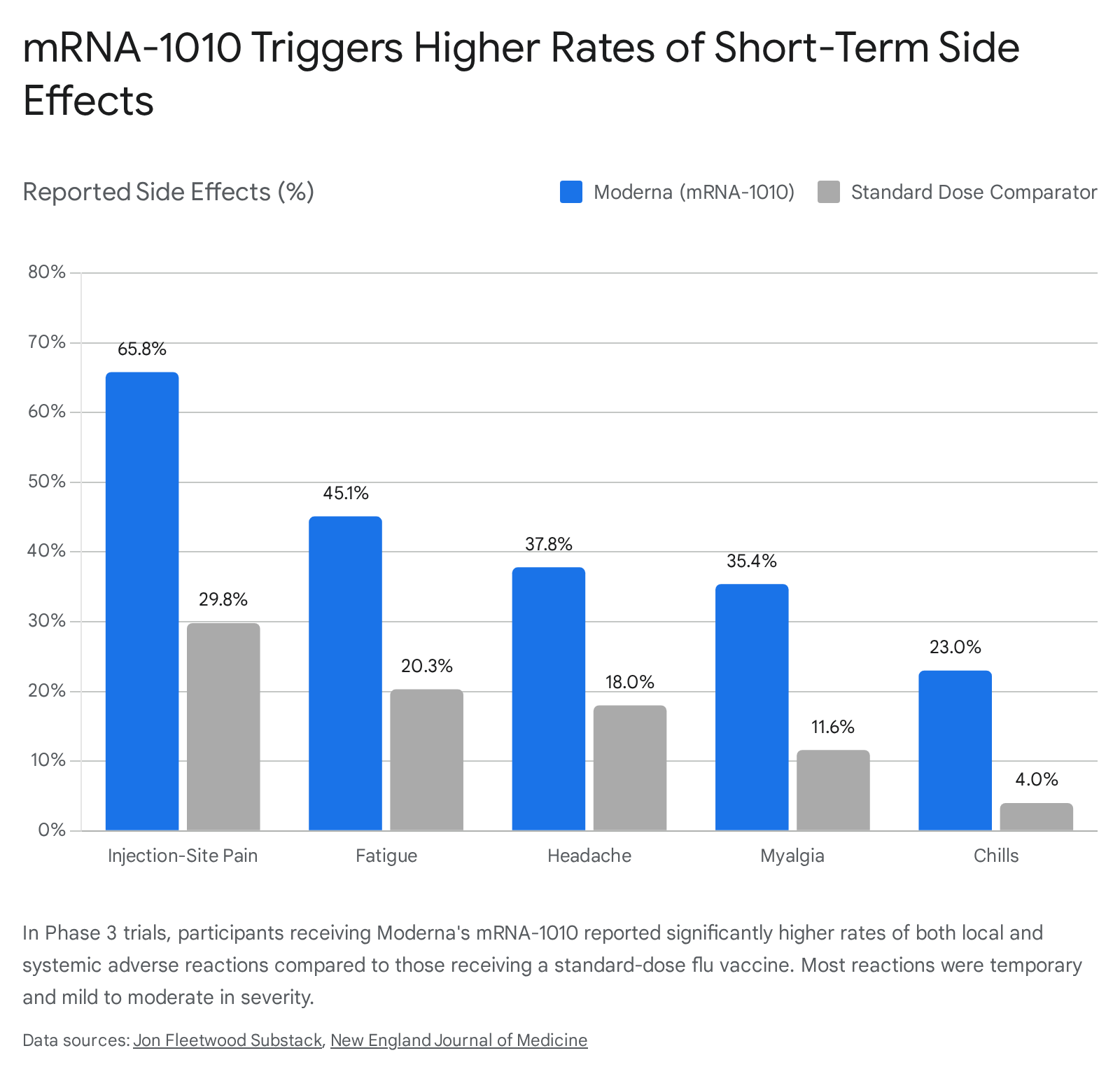
While the vast majority of these solicited adverse reactions were classified as Grade 1 or Grade 2 (mild to moderate and transient), the trial specifically measured "Grade 3" reactions. A Grade 3 reaction is classified as severe enough to actively interfere with a person's ability to perform normal daily activities, such as working or routine movement 24.
In the P304 trial, 6.4% of mRNA-1010 recipients experienced a Grade 3 solicited adverse reaction, compared to just 1.0% of standard-dose flu shot recipients 192426. This indicates that approximately one in sixteen people who received the mRNA flu shot experienced a severe, day-disrupting short-term reaction, compared to roughly one in one hundred for the traditional shot 1924.
Despite the high rates of short-term reactogenicity, the long-term safety profile proved highly favorable. Serious adverse events (SAEs) extending beyond immediate vaccine reactogenicity were infrequent and statistically similar between the two cohorts, reported in 2.2% of mRNA-1010 recipients and 1.9% of standard-dose recipients 1626. Furthermore, an independent Cardiac Event Adjudication Committee (CEAC) assessed the trial data and found no cases of acute myocarditis or pericarditis linked to the mRNA-1010 vaccine within the conservative 42-day risk window post-vaccination 926.
A Regulatory Rollercoaster: The Refusal-to-File Drama
Despite meeting all pre-specified primary clinical endpoints, Moderna's journey through the FDA's regulatory apparatus in early 2026 was highly turbulent, shedding light on the shifting political and operational dynamics within federal health agencies.
On February 10, 2026, the FDA's Center for Biologics Evaluation and Research (CBER) issued a highly unusual "Refusal-to-File" (RTF) letter for Moderna's Biologics License Application (BLA) 171819. An RTF is an administrative mechanism usually reserved for applications that are grossly incomplete or fundamentally flawed, indicating that the agency refuses to even begin reviewing the data 1820.
The "Standard of Care" Comparator Dispute
Crucially, the FDA's refusal was not based on any underlying concerns regarding the safety, immunogenicity, or efficacy of the vaccine itself 181921. Instead, CBER Director Dr. Vinay Prasad rejected the application solely based on the choice of the comparator vaccine used in the P304 Phase 3 trial 18192223.
In the P304 trial, which enrolled adults aged 50 and older, Moderna compared its mRNA vaccine against a standard-dose licensed seasonal influenza vaccine 1718. However, for adults aged 65 and older, the CDC's Advisory Committee on Immunization Practices (ACIP) preferentially recommends "enhanced" vaccines - such as high-dose (Fluzone HD), adjuvanted (FLUAD), or recombinant (Flublok) vaccines - because the aging immune system requires a stronger stimulus to generate adequate protection 1718.
The FDA argued that by using a standard-dose vaccine as the baseline for participants over 65, Moderna had failed to conduct an "adequate and well-controlled" study because the control arm did not reflect the "best-available standard of care" recommended for that specific age demographic 182123. The FDA claimed this approach essentially exposed senior participants in the control group to an increased risk of severe illness by providing them with a substandard preventative measure 24.
Moderna mounted an immediate and public defense. The biotechnology company released communications demonstrating that the FDA had reviewed and explicitly cleared the Phase 3 study design in April 2024, nearly 18 months before the RTF was issued 181924. During those early consultations, the FDA had recommended using a high-dose comparator for seniors but ultimately agreed in writing that using a licensed standard-dose vaccine across the entire trial population would be "acceptable" 181921. Moderna further argued that Title 21 of the Code of Federal Regulations governing adequate and well-controlled clinical trials contains no legal mandate requiring comparators to meet a newly defined "best-available standard of care" 1819.
Contextualizing the FDA Tumult
This regulatory impasse did not occur in a vacuum. Early 2026 was marked by immense upheaval at the highest levels of the US health apparatus. Under the new leadership of Health and Human Services (HHS) Secretary Robert F. Kennedy Jr. - a prominent critic of vaccines who specifically targeted mRNA technology and slashed $500 million in mRNA-related federal contracts - the FDA faced intense ideological and structural pressure 16223625.
During this period, Secretary Kennedy replaced every member of the CDC's ACIP panel, and FDA Commissioner Marty Makary abruptly resigned in May 2026 amidst the ongoing turmoil 1838. The RTF issued to Moderna was widely viewed by industry analysts as a manifestation of this heightened, highly unpredictable regulatory scrutiny 223839.
The Swift Reversal and Bifurcated Compromise
Facing immense public pushback and allegations of regulatory inconsistency, the FDA granted Moderna an emergency Type A meeting 172326. Just one week after issuing the RTF, the FDA reversed course and agreed to accept an amended application under a newly negotiated, bifurcated regulatory strategy 172236.
To bridge the gap between Moderna's trial design and the FDA's demand for high-dose comparators in seniors, the two parties split the approval pathway based on age 1727.
| Target Age Demographic | Revised FDA Approval Pathway | Specific Conditions & Requirements |
|---|---|---|
| Adults 50 to 64 Years | Full, Traditional Approval | Evaluated primarily on the P304 efficacy data against standard-dose comparators, which aligns with existing standards of care for this age group 1727. |
| Adults 65 and Older | Accelerated Approval | Granted conditionally based on surrogate endpoints, requiring Moderna to fulfill a post-marketing commitment to conduct an entirely new outcomes study explicitly comparing mRNA-1010 against a high-dose vaccine in seniors 17233627. |
Following this compromise, the FDA formally accepted the Biologics License Application and assigned a Prescription Drug User Fee Act (PDUFA) action date of August 5, 2026 17232627.
The Final Hurdle: The June 18 VRBPAC Meeting
Before the FDA renders its final decision on the August 5 PDUFA date, mFLUSIVA faces a critical public evaluation. The FDA has scheduled a meeting of the Vaccines and Related Biological Products Advisory Committee (VRBPAC) for June 18, 2026 282930.
This panel, comprised of independent immunologists, infectious disease experts, and biostatisticians, acts as a vital scientific sounding board for the agency 730. During the open session, the committee will scrutinize Moderna's Phase 3 clinical data, directly weighing the 26.6% relative efficacy benefit against the pronounced increase in short-term reactogenicity and Grade 3 adverse events 7242639.
The meeting carries immense precedent-setting weight. While the FDA routinely convened VRBPAC during the COVID-19 pandemic, this June 2026 meeting represents the first time an advisory panel will formally debate the long-term clinical viability, safety profile, and broad public health merits of applying mRNA technology to a seasonal, annually recurring virus outside of a public health emergency 739. While VRBPAC recommendations are non-binding, the FDA traditionally adheres closely to the panel's consensus votes 730.
The Competitive Landscape: Pfizer, Sanofi, and GSK
Moderna is not operating in a vacuum; the race to develop next-generation influenza vaccines has attracted vast investments from the world's largest pharmaceutical companies, though the results have been highly mixed.
Sanofi Abandons Standalone mRNA Flu Shots
The most notable shift in the competitive landscape occurred in early 2026 when Sanofi - a titan in the traditional influenza vaccine market - abruptly scrapped its seasonal mRNA flu vaccine program 142531.
Following its $3.2 billion acquisition of mRNA-specialist Translate Bio in 2021, Sanofi advanced a hexavalent mRNA-based seasonal shot (SP0237) into Phase 1/2 trials 142531. However, mirroring the struggles faced by early iterations of Moderna's candidate, Sanofi's lipid nanoparticles performed exceptionally well against influenza A but failed to consistently neutralize influenza B strains 1446. Ultimately, Sanofi concluded the technology was not mature enough to displace its highly successful, traditional offerings (Fluzone High-Dose and Flublok), effectively ceding the standalone mRNA seasonal market to its rivals 1446. Sanofi is now restricting its mRNA flu research strictly to a Phase 1/2 trial for a potential H5 avian flu pandemic vaccine 143146.
Pfizer and GSK Press Forward
Pfizer and its partner BioNTech are aggressively pursuing their own mRNA flu vaccine. In a recent 18,000-person global Phase 3 trial, Pfizer's mRNA vaccine demonstrated an impressive 34.5% higher relative efficacy at preventing lab-confirmed flu compared to the licensed inactivated vaccine Fluzone 312. However, Pfizer's data revealed the same inherent platform vulnerabilities seen across the industry: weaker antibody responses against influenza B strains and a significantly higher rate of systemic side effects, such as fever and chills 312.
Meanwhile, GSK, leveraging technology from its €400 million partnership with CureVac, recently reported highly positive Phase 2 results for its multivalent mRNA candidate, successfully eliciting positive immune responses against both A and B strains across younger and older adults 47. GSK is aiming to file for regulatory approval in 2026 with a potential market launch in 2027, positioning it to challenge Moderna and Pfizer rapidly 47.
The Next Frontier: Combination Vaccines
While the approval of mFLUSIVA represents a technological milestone, industry analysts and pharmaceutical executives increasingly view standalone mRNA flu shots as a transitional stepping stone. The ultimate endgame is the deployment of combination vaccines that simultaneously target multiple respiratory pathogens in a single annual injection.
Moderna is already well advanced in this arena. The company's mRNA-1083 candidate combines the seasonal influenza components of mRNA-1010 with mRNA-1283, a next-generation COVID-19 vaccine targeting recent Omicron subvariants like XBB.1.5 133249.
In a Phase 3 trial (Duo P301) involving over 8,000 adults aged 50 and older, a single dose of mRNA-1083 was tested against the co-administration of two separate, best-in-class licensed vaccines (Sanofi's Fluzone HD and Moderna's own Spikevax) 11133233. The results demonstrated that the combination shot met all non-inferiority criteria and actually elicited statistically higher immune responses against three influenza strains and SARS-CoV-2 compared to receiving the two traditional shots separately 13324933.
With "vaccine fatigue" driving down compliance rates globally, health systems view combination vaccines as the primary strategy to reduce administrative burdens on pharmacies and ensure broad population protection against overlapping winter viruses 133251. Moderna plans to file for regulatory approval of its combo jab in the summer of 2026, aiming for a 2027 market entry 49.
Fall 2026: Pharmacy Availability and Logistics
If the FDA meets its August 5 PDUFA deadline and issues an approval, Moderna intends to launch mFLUSIVA immediately for the 2026 - 2027 Northern Hemisphere flu season 2627.
From a logistical standpoint, an August approval aligns well with standard public health recommendations. The CDC and state health departments generally advise adults to receive their annual flu vaccinations in September and October, ensuring peak immunity aligns with the historical peak of winter viral transmission 2334.
However, Moderna will face fierce commercial headwinds from incumbent manufacturers. Companies like GSK and Sanofi traditionally secure massive pre-booking contracts early in the year and begin physically shipping their egg-based and recombinant doses to pharmacies and clinics by July or early August 3554. For the 2025 - 2026 season, vaccine manufacturers projected supplying up to 154 million doses to the US market 55. Moderna will have to execute a flawless, highly accelerated supply chain pivot to distribute its LNP-encapsulated mRNA doses into a market saturated with established brands mere weeks after receiving FDA clearance.
Bottom line
The FDA is on track to approve Moderna's mFLUSIVA by August 5, 2026, representing a pivotal shift away from antiquated egg-based vaccine manufacturing. The robust Phase 3 clinical data demonstrates that the mRNA vaccine offers superior protection against the flu compared to standard-dose vaccines, significantly reducing absolute infection rates across older adult populations. However, this enhanced efficacy comes with a pronounced increase in short-term side effects - such as severe fatigue, chills, and injection-site pain - that patients must weigh against the benefits. Provided it clears a critical advisory committee review in June, mFLUSIVA will initiate a new era of rapid-response mRNA therapeutics, setting the stage for highly potent combination respiratory vaccines in the years to follow.