Key Biotech FDA Decisions from August to December 2026
The second half of 2026 features a critical sequence of FDA Prescription Drug User Fee Act (PDUFA) target action dates that could introduce transformative therapies across oncology, rare diseases, and psychiatry. Notable anticipated approvals include Nuvalent's next-generation targeted lung cancer therapies, Capricor Therapeutics' cellular therapy for Duchenne muscular dystrophy, and potentially the revolutionary weight-loss combination CagriSema from Novo Nordisk. Investors, clinicians, and patients are closely monitoring these regulatory milestones, as the resulting decisions will heavily influence clinical treatment paradigms and biotechnology market valuations into the 2030s.
Navigating the FDA Regulatory Labyrinth
To accurately track and anticipate movements within the biotechnology sector, one must understand the intricate mechanisms that dictate drug approvals in the United States. The Prescription Drug User Fee Act (PDUFA), originally enacted in 1992, authorizes the U.S. Food and Drug Administration (FDA) to collect fees from drug manufacturers to fund the medical product approval process. In exchange, the FDA commits to specific target action dates by which it aims to complete its review of a New Drug Application (NDA) or a Biologics License Application (BLA) 12.
The regulatory pathway leading up to a PDUFA date is frequently the subject of public and market misunderstanding. A common misconception is that the FDA acts as an overly burdensome bureaucratic hurdle that arbitrarily delays life-saving medications, or conversely, that the agency rubber-stamps experimental drugs based on minimal Phase 1 or Phase 2 research 335. In reality, regular approvals based on rigorous, late-stage efficacy data far exceed accelerated approvals. Between 2016 and 2019, for instance, there were 115 regular approvals versus only 39 accelerated approvals for new molecular entities 3. The FDA evaluates extensive, multi-phase clinical trial data to balance clinical efficacy with long-term safety profiles, and the agency relies heavily on post-market surveillance to ensure that rare adverse events are addressed swiftly 354.
Furthermore, critics occasionally claim that the FDA is slower than foreign agencies, such as the European Medicines Agency (EMA), but historical data suggests the FDA frequently evaluates and approves drugs at a faster rate than its global counterparts 4. To facilitate this, the FDA utilizes several expedited pathways designed to speed the development of promising therapies for serious illnesses.
Expedited Pathways and Rolling Reviews
When an investigational drug treats a serious or life-threatening condition and addresses an unmet medical need - defined as providing therapy where none currently exists or offering a significant advantage over available treatments - the FDA can grant special designations to accelerate its path to market 25.
Understanding the distinctions between these regulatory mechanisms is crucial for evaluating a drug's developmental timeline:
| Regulatory Designation | Primary Benefit and Function |
|---|---|
| Fast Track | Facilitates more frequent meetings and written correspondence with the FDA. It ensures early and consistent communication throughout the development process to resolve issues quickly 25. |
| Breakthrough Therapy | Granted when preliminary clinical evidence indicates the drug may demonstrate substantial improvement over existing therapies. It provides all Fast Track benefits plus intensive FDA guidance on efficient drug development and organizational involvement from senior FDA management 2. |
| Priority Review | Shortens the FDA's targeted review time from the standard 10 months to just 6 months. This is typically granted to drugs that, if approved, would provide significant improvements in the safety or effectiveness of treatment 29. |
| Accelerated Approval | Allows for approval based on a surrogate endpoint (a marker thought to predict clinical benefit) rather than a direct measure of clinical benefit, expediting access for serious conditions. Post-marketing confirmatory trials are required 26. |
| Rolling Review | Allows sponsors to submit individually completed sections of an NDA or BLA for FDA review before the entire application is finished. The FDA reviews the modules incrementally, significantly compressing the final review timeline 257. |
The Reality of Review Extensions and Major Amendments
A PDUFA date represents a target goal, not an ironclad, legally binding guarantee. The FDA can, and frequently does, extend these dates when new information is submitted late in the review cycle.
A prominent example of this mechanism in action occurred with Travere Therapeutics and its drug Filspari (sparsentan). Originally, Travere held a PDUFA target action date of January 13, 2026, for the full approval of Filspari in the treatment of focal segmental glomerulosclerosis (FSGS) 128. Late in the review process, the FDA issued information requests to further characterize the drug's clinical benefit 129. When Travere submitted the requested responses, the FDA classified the new data as a "Major Amendment" to the application 910. By standard protocol, a Major Amendment automatically extends the review period by three months to allow regulators adequate time to evaluate the new data. Consequently, Filspari's PDUFA date was pushed to April 13, 2026 1016. Regulatory extensions like this are standard procedure and do not necessarily indicate impending rejection or the discovery of new safety concerns, though they frequently trigger volatility in biotechnology stock valuations 910.
Late 2026 Biotech FDA Calendar Summary
The latter half of 2026 is densely packed with regulatory decisions that span preventative vaccines, targeted cancer therapies, and treatments for ultra-rare genetic disorders. The table below provides a comprehensive summary of the publicly confirmed PDUFA target action dates scheduled from August through December 2026.
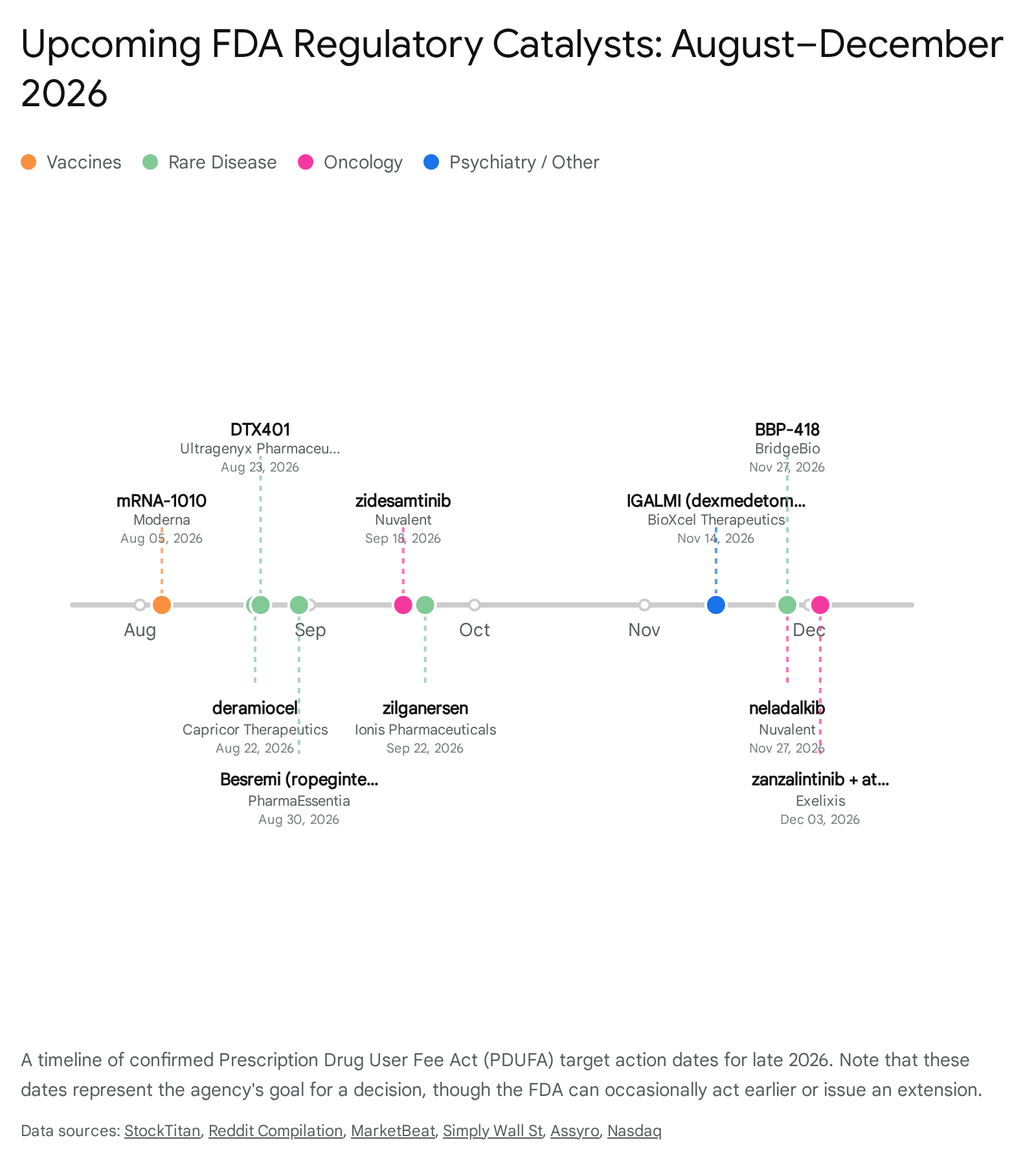
| PDUFA Date (2026) | Company | Drug Candidate | Target Indication / Therapeutic Use |
|---|---|---|---|
| August 5 | Moderna | mRNA-1010 | Seasonal influenza illness |
| August 22 | Capricor Therapeutics | Deramiocel | Duchenne muscular dystrophy (DMD) cardiomyopathy |
| August 23 | Ultragenyx | DTX401 | Glycogen storage disease type Ia |
| August 30 | PharmaEssentia | Besremi | Essential thrombocythemia |
| September 18 | Nuvalent | Zidesamtinib | ROS1-positive non-small cell lung cancer (NSCLC) |
| September 22 | Ionis Pharmaceuticals | Zilganersen | Alexander disease |
| November 14 | BioXcel Therapeutics | IGALMI (BXCL501) | Agitation in bipolar disorder/schizophrenia (at-home use) |
| November 27 | Nuvalent | Neladalkib | ALK-positive non-small cell lung cancer (NSCLC) |
| November 27 | BridgeBio | BBP-418 | Limb-girdle muscular dystrophy type 2I/R9 |
| December 3 | Exelixis | Zanzalintinib + Atezolizumab | Metastatic colorectal cancer (mCRC) |
(Data aggregated from corporate SEC filings, FDA public dockets, and clinical trial registries 11181920212223. Note that Novo Nordisk's CagriSema is anticipated for a decision in late 2026 or early 2027, though an exact public PDUFA calendar day has not been uniformly declared 24.)
August 2026: Vaccines, Cell Therapies, and Rare Diseases
The late summer regulatory calendar kicks off with high-stakes decisions impacting both widespread public health initiatives and highly specialized rare disease communities.
Moderna's mRNA-1010 for Seasonal Influenza (August 5)
Moderna is seeking regulatory approval for its seasonal influenza vaccine candidate, mRNA-1010, marking a critical test of the company's ability to successfully expand its messenger RNA (mRNA) technology platform beyond the COVID-19 pandemic 2312.
The public health burden of seasonal influenza remains vast and economically taxing. According to the U.S. Centers for Disease Control (CDC), seasonal flu-related hospitalizations and outpatient visits reached a 15-year high during the 2024 - 2025 season 2326. More than 545,000 Americans were hospitalized due to flu-related illness during that period, leading to substantial direct and indirect medical costs, as well as widespread disruption to daily life 26. Current standard-of-care flu vaccines often struggle with efficacy mismatch due to the long lead times required for egg-based viral cultivation. Moderna argues that its mRNA-based seasonal flu vaccine has the potential to more precisely match circulating viral strains because of the rapid manufacturing capabilities inherent to mRNA technology 23.
The NDA for mRNA-1010 is backed by robust Phase 3 clinical data, specifically the P304 study, which evaluated the relative vaccine efficacy (rVE) of mRNA-1010 against influenza illness compared to a licensed standard-dose seasonal influenza vaccine in adults aged 50 years and older 23. The results, published in the New England Journal of Medicine, demonstrated an overall relative vaccine efficacy of 26.6% versus the standard-dose active comparator 26. The vaccine showed particularly strong strain-specific relative efficacy against A/H1N1 (29.6% rVE) and B/Victoria (29.1% rVE) 26. Importantly, subgroup analyses revealed consistent efficacy across age groups and risk factors. In the highly vulnerable demographic of participants aged 65 and older, mRNA-1010 maintained a robust 27.4% rVE 26.
The FDA has assigned mRNA-1010 a PDUFA goal date of August 5, 2026 231226. The vaccine is concurrently under regulatory review in Europe, Canada, and Australia, with potential global approvals expected to begin cascading in late 2026 231213. If the FDA grants approval on or before the target date, Moderna anticipates incorporating the vaccine into commercial rollouts in time for the upcoming Northern Hemisphere flu seasons 1226.
Capricor Therapeutics' Deramiocel for DMD Cardiomyopathy (August 22)
Capricor Therapeutics is approaching a high-stakes, binary catalyst on August 22, 2026, for deramiocel, an investigational cell and exosome-based therapy indicated for the treatment of Duchenne muscular dystrophy (DMD) cardiomyopathy 212829.
Duchenne muscular dystrophy is a severe, X-linked genetic disorder characterized by the progressive degeneration of skeletal, respiratory, and cardiac muscles 29. The disease is caused by a genetic mutation that prevents the body from producing functional dystrophin, a key structural protein required to maintain muscle cell integrity 29. Affecting approximately 15,000 individuals in the United States - primarily boys - the disease inevitably leads to wheelchair dependency, respiratory failure, and severe cardiomyopathy, the latter of which is the leading cause of mortality in older DMD patients 29. While deramiocel is not a cure for the underlying genetic defect, the therapy is designed to slow the relentless destruction of cardiac tissue, thereby preserving heart function, prolonging lifespan, and improving quality of life 29.
The regulatory journey for deramiocel has been fraught with drama and sudden reversals. The FDA had previously issued a Complete Response Letter (CRL) for the therapy, stalling its path to commercialization 2830. However, in a major victory for Capricor, the company engaged in discussions with the FDA and presented compelling clinical study reports from the pivotal Phase 3 HOPE-3 trial 30. Satisfied with the new data, the FDA lifted the CRL without requiring the company to conduct expensive and time-consuming new clinical studies 2830. The agency subsequently allowed a Class 2 resubmission of the BLA and set a near-term PDUFA date of August 22, 2026 2830.
The clinical data underpinning the resubmission is highly encouraging. The Phase 3 HOPE-3 trial demonstrated that treatment with deramiocel slowed overall disease progression by 54%, as measured by the Performance of the Upper Limb (PUL) assessment 3014. Even more critically for the specific cardiomyopathy indication, the therapy yielded a 91% improvement in Left Ventricular Ejection Fraction (LVEF), a gold-standard measurement of cardiac pumping efficiency 30.
Beyond the clinical implications, an FDA approval would be a massive financial boon for Capricor. The agency has granted deramiocel Priority Review status, making the company eligible to receive a highly coveted Priority Review Voucher upon approval 282930. These vouchers, which can be sold to larger pharmaceutical companies to expedite the review of other blockbuster drugs, typically command prices exceeding $100 million in the open market, providing non-dilutive capital to biotech firms 2830. Despite ongoing legal friction regarding a U.S. distribution agreement with partner NS Pharma, Capricor maintains a strong cash position of approximately $318 million, providing a financial runway through 2027 to navigate the commercial launch 293014.
Additional August Milestones: Ultragenyx and PharmaEssentia
Rounding out the busy month of August, Ultragenyx faces an August 23, 2026, PDUFA date for DTX401, an investigational adeno-associated virus (AAV) gene therapy targeting Glycogen storage disease type Ia, a rare metabolic disorder 21. Shortly thereafter, on August 30, PharmaEssentia anticipates an FDA decision on a supplemental Biologics License Application (sBLA) for Besremi (ropeginterferon alfa-2b). The company is seeking to expand the drug's label for the treatment of essential thrombocythemia, a rare blood cancer characterized by the overproduction of platelets 18.
September 2026: The Rise of Next-Generation TKIs
September 2026 is dominated by decisions surrounding precision oncology and targeted neurology, with a specific focus on the next generation of tyrosine kinase inhibitors (TKIs).
Nuvalent's Zidesamtinib for ROS1-Positive NSCLC (September 18)
Nuvalent is awaiting a September 18, 2026, PDUFA decision for zidesamtinib, a novel ROS1-selective inhibitor aimed at adult patients with locally advanced or metastatic ROS1-positive non-small cell lung cancer (NSCLC) who have previously received at least one prior ROS1 tyrosine kinase inhibitor 221516.
Tyrosine kinase inhibitors have revolutionized the treatment of genetically driven lung cancers, but patients inevitably develop resistance to first- and second-generation therapies. Zidesamtinib was specifically engineered from the ground up to overcome the limitations associated with earlier TKIs. The drug's NDA is anchored by pivotal data from the global ARROS-1 Phase 1/2 clinical trial 223417.
The clinical data presented at the American Association for Cancer Research (AACR) and the American Society of Clinical Oncology (ASCO) meetings highlighted two critical advantages of zidesamtinib 153417. First, the drug possesses high central nervous system (CNS) penetrance. Lung cancer frequently metastasizes to the brain, where many older drugs cannot reach due to the blood-brain barrier. Zidesamtinib successfully yielded intracranial complete responses in patients with severe CNS disease 1517. Second, zidesamtinib demonstrated significant clinical activity against tumors harboring the notoriously difficult ROS1 G2032R resistance mutation 3417. The drug proved highly effective even in heavily pre-treated patients who had progressed on newer, highly potent TKIs like repotrectinib and taletrectinib, proving that ROS1-positive NSCLC tumors remain dependent on the ROS1 pathway and can be targeted sequentially if the inhibitor is designed correctly 1517.
Nuvalent is heavily invested in its commercial infrastructure ahead of a potential U.S. launch in late 2026 1617. The company is also planning to submit additional data to the FDA in the second half of 2026 to support a label expansion into TKI-naïve (first-line) patients, seeking to move zidesamtinib earlier in the treatment paradigm 17.
Ionis Pharmaceuticals' Zilganersen for Alexander Disease (September 22)
Outside the oncology sphere, Ionis Pharmaceuticals holds a Priority Review PDUFA target action date of September 22, 2026, for zilganersen 1836. Zilganersen is an investigational antisense oligonucleotide (ASO) therapy intended for the treatment of children and adults with Alexander disease (AxD), an ultra-rare, progressive, and fatal leukodystrophy 36. If approved, it would mark a significant milestone in the treatment of rare neurological disorders using RNA-targeted technologies.
October and November 2026: Psychiatry, NASH, and More TKIs
The late autumn calendar introduces regulatory decisions that could reshape outpatient psychiatric care and expand therapies for epidemic-scale metabolic diseases.
BioXcel's IGALMI for At-Home Psychiatric Agitation (November 14)
BioXcel Therapeutics has secured a November 14, 2026, PDUFA target action date for a supplemental New Drug Application (sNDA) that seeks to radically expand the label for its drug IGALMI (dexmedetomidine, internally referred to as BXCL501) 111838.
IGALMI, a selective alpha-2 adrenergic receptor agonist delivered via an orally dissolving sublingual film, is currently approved by the FDA for the acute treatment of mild, moderate, or severe agitation in patients with schizophrenia or bipolar I or II disorder 3819. However, the current label restricts its administration strictly to supervised healthcare settings, such as hospitals and psychiatric emergency departments 183819.
The sNDA seeks to allow IGALMI to be self-administered by patients at home, without the supervision of a healthcare provider 183840. This represents a significant paradigm shift in psychiatric care. Currently, there are no FDA-approved options for treating severe agitation episodes in a non-supervised outpatient setting 1819. When agitation escalates at home, it frequently results in traumatic emergency room visits, forced sedation, or hospitalization. Providing a rapid-onset, self-administered calming agent could grant unprecedented autonomy to patients managing chronic psychiatric conditions 1940.
The application relies heavily on data from the pivotal Phase 3 SERENITY At-Home safety trial 1838. The double-blind, placebo-controlled 12-week study evaluated patients self-administering the 120 mcg dose of BXCL501 during agitation episodes 3840. During the trial, more than 2,400 distinct episodes of agitation were treated in the outpatient setting 40. The drug successfully met its primary endpoint, proving to be well-tolerated across repeat dosing with no discontinuations required due to tolerability issues 40. Furthermore, secondary analyses demonstrated that BXCL501 reduced agitation versus placebo across all baseline severities, with the most pronounced treatment effect observed in patients experiencing severe agitation episodes 11. BioXcel executives estimate that opening the at-home market vastly increases the total addressable market for the drug 40.
Nuvalent's Neladalkib for ALK-Positive NSCLC (November 27)
Following its September regulatory date for zidesamtinib, Nuvalent faces a second major PDUFA milestone on November 27, 2026, for its drug neladalkib 20. This NDA, which has been granted Priority Review, targets the treatment of patients with advanced ALK-positive non-small cell lung cancer (NSCLC) who have progressed after prior TKI therapies 2016.
Neladalkib is an investigational ALK-selective inhibitor. Much like zidesamtinib, it is designed to overcome specific resistance mutations and penetrate the central nervous system. Data from the global ALKOVE-1 Phase 1/2 clinical trial supports the application 2022. The drug has received Breakthrough Therapy designation from the FDA, acknowledging its potential to substantially improve outcomes for patients who have exhausted existing therapies 20. Analysts note that securing approvals for both zidesamtinib and neladalkib within months of each other would swiftly transform Nuvalent from a clinical-stage outfit into a dual-asset commercial oncology company 1617. Supported by a robust cash position of $1.3 billion, the company projects its financial runway extends into 2029 16.
Madrigal's Rezdiffra Label Expansion (November 27)
Also slated for November 27, 2026, is an FDA decision regarding Madrigal Pharmaceuticals and its drug Rezdiffra (resmetirom) 20. Rezdiffra recently broke ground as the very first medication approved in the United States (and subsequently in Europe) for the treatment of metabolic dysfunction-associated steatohepatitis (MASH) - historically referred to as nonalcoholic steatohepatitis (NASH) 4120.
Madrigal is currently seeking to expand Rezdiffra's label based on new analyses from its Phase 3 MAESTRO program and emerging real-world evidence 2043. The therapy treats adults with noncirrhotic MASH accompanied by moderate to advanced liver fibrosis 4321. Recent data presented at the European Association for the Study of the Liver (EASL) Congress highlighted that Rezdiffra not only resolves liver inflammation but significantly improves atherogenic lipid profiles associated with cardiovascular risk, including reductions in LDL-C, ApoB, and Lp(a) 20. As the drug launch progresses, achieving broad label definitions will be critical for Madrigal to fend off looming competition from blockbuster GLP-1 weight-loss drugs that are also being evaluated for liver disease 41.
BridgeBio's BBP-418 for Limb-Girdle Muscular Dystrophy (November 27)
BridgeBio is anticipating an FDA decision on November 27, 2026, for BBP-418, an investigational oral therapy for the treatment of individuals living with limb-girdle muscular dystrophy type 2I/R9 (LGMD2I/R9) 20. The NDA has been granted Priority Review, and notably, the FDA informed the company that it does not currently plan to hold an advisory committee meeting to discuss the application, which often signals a smooth review process 20.
December 2026: Combinatorial Oncology and Potential Megablockbusters
The year concludes with regulatory actions surrounding combination therapies in solid tumors and the highly anticipated arrival of next-generation obesity medications.
Exelixis's Zanzalintinib for Metastatic Colorectal Cancer (December 3)
Exelixis closes out the 2026 calendar year with a December 3 standard review PDUFA date for zanzalintinib (formerly XL092), utilized in combination with the immune checkpoint inhibitor atezolizumab (Tecentriq) 192223. This combination therapy is targeted at adult patients with metastatic colorectal cancer (mCRC) who have previously exhausted standard fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapies, and - if they harbor a RAS wild-type mutation - prior anti-EGFR therapies 192224.
Zanzalintinib is a novel, next-generation oral multi-kinase inhibitor that specifically targets MET, VEGFR, and the TAM family of kinases (TYRO3, AXL, and MER) 2425. These kinase pathways are heavily implicated in tumor cell proliferation, metastasis, angiogenesis, drug resistance, and the evasion of antitumor immunity 25. By inhibiting these specific targets, zanzalintinib is theorized to favorably modulate the immunosuppressive tumor microenvironment, thereby enhancing the efficacy of atezolizumab, a standard immunotherapy 2224.
The regulatory submission is supported by compelling data from the pivotal Phase 3 STELLAR-303 trial 2224. STELLAR-303 made clinical history as the first Phase 3 study of an immune checkpoint inhibitor-based combination to demonstrate a statistically significant overall survival (OS) improvement in this specific, heavily pre-treated patient population - crucially, patients whose tumors are microsatellite stable (MSS) or mismatch repair proficient, a group historically resistant to immunotherapy 2224.
In the intent-to-treat population of 901 patients, the zanzalintinib and atezolizumab combination extended the median overall survival to 10.9 months, compared to 9.4 months for patients treated with the standard-of-care regorafenib (Hazard Ratio 0.80; p=0.0045) 26. The combination also significantly improved progression-free survival (3.7 months vs 2.0 months) and resulted in a higher disease control rate (54% vs 41%) 24. An approval would establish zanzalintinib as a crucial chemotherapy-free option for refractory metastatic colorectal cancer, and Exelixis is concurrently pursuing pivotal Phase 3 trials (STELLAR-304 and STELLAR-316) to expand the drug into renal cell carcinoma and earlier stages of CRC 2223242526.
Novo Nordisk's CagriSema for Obesity (Late 2026 / Early 2027)
While it does not currently have a publicly finalized, specific calendar date beyond "late 2026" or early 2027, Novo Nordisk's CagriSema requires intense scrutiny, as it is widely projected to be the most commercially significant drug reviewed during this cycle 2427.
Novo Nordisk submitted the NDA for CagriSema to the FDA in December 2025 2452. The company is seeking approval for chronic weight management in adults with obesity or overweight who have at least one weight-related comorbidity (such as type 2 diabetes, obstructive sleep apnea, or heart disease) 27522829.
CagriSema represents a conceptual leap beyond the current generation of weight-loss blockbusters. It is a fixed-dose combination of semaglutide (a GLP-1 receptor agonist, the active ingredient in Wegovy and Ozempic) and cagrilintide (a novel, long-acting amylin analog) 27282930. Both components are dosed at 2.4 mg and delivered simultaneously via a single, once-weekly subcutaneous injection 2856. By targeting two entirely different hormonal pathways regulating hunger, satiety, and metabolic rate, CagriSema achieves synergistic efficacy far exceeding current monotherapies 2830.
The clinical data is extraordinary. In the Phase 3 REDEFINE 1 trial, patients treated with CagriSema achieved a staggering average weight loss of 22.7% over 68 weeks 52285657. For context, this drastically outperformed the 16.1% average reduction seen with semaglutide alone, and the 2.3% reduction seen in the placebo cohort 2856. The consistency of the response was equally notable: 91.9% of participants taking CagriSema achieved at least a 5% reduction in body weight, and an unprecedented 54% of treated patients lost enough weight to transition entirely below the clinical BMI threshold defining obesity 2956.
Furthermore, data from the REIMAGINE 2 trial demonstrated that CagriSema achieved superior HbA1c reductions (up to 1.91 percentage points) in patients with type 2 diabetes compared to semaglutide alone, indicating powerful glycemic control benefits alongside weight loss 58.
Industry analysts predict that CagriSema will make an immediate and disruptive impact on the market, with projections estimating peak annual sales reaching $17.2 billion by 2032 273057. Novo Nordisk is also exploring a "CagriSema Forte" high-dose version incorporating 7.2 mg of semaglutide, aiming to push weight loss ceilings even higher 242728. Under a standard 10-to-12-month regulatory review timeline stemming from its December 2025 filing, the FDA's final decision is anticipated in late 2026, though some analysts hedge that the official commercial launch could slip into early 2027 242729.
Broad Context: Bispecific Antibodies and Emerging Innovations
As Exelixis and Nuvalent approach their late-2026 PDUFA dates, they enter an oncology market that is being rapidly disrupted by the success of bispecific antibodies - a trend that contextualizes the urgency of these FDA reviews.
For example, ivonescimab, a novel first-in-class bispecific antibody developed by Akeso and partnered with Summit Therapeutics, targets both PD-1 (inhibiting immune checkpoint evasion) and VEGF-A (inhibiting tumor angiogenesis) 313233. The drug is generating unprecedented clinical data in lung cancer trials. In the Phase 3 HARMONi-A trial, ivonescimab combined with chemotherapy achieved a statistically significant overall survival benefit (a median of 16.8 months versus 14.1 months) in patients with EGFR-mutated NSCLC who had progressed on prior TKIs 3435. Similarly, the HARMONi-6 and HARMONi-2 trials have shown ivonescimab significantly improving progression-free survival when tested head-to-head against standard immunotherapies like tislelizumab and pembrolizumab 3233. As bispecifics raise the standard of care, newly approved targeted TKIs and combinations will have to navigate an increasingly competitive and efficacious treatment landscape 33.
Elsewhere, innovative delivery mechanisms and platforms continue to navigate the FDA calendar. MannKind Corporation anticipates an FDA decision by May 29, 2026, for a pediatric label expansion of Afrezza, an inhaled insulin powder aiming to become the first needle-free insulin option for children and adolescents aged 4 to 17 with type 1 or type 2 diabetes 19161820. Additionally, Inovio Pharmaceuticals is preparing to submit a BLA in mid-2025 for INO-3107, an investigational DNA medicine targeting recurrent respiratory papillomatosis (RRP) 3665. RRP is a debilitating disease characterized by benign growths in the respiratory tract caused by HPV-6 and HPV-11 3666. INO-3107 utilizes engineered DNA to trigger an antigen-specific T-cell response, and extension studies have shown it reduces the mean number of required airway surgeries from 4.1 to 0.9 per year 36653768. While its PDUFA date will likely fall outside the 2026 window, its Breakthrough Therapy designation highlights the FDA's willingness to accelerate novel mechanisms of action for chronic, rare morbidities 1936.
Bottom line
The FDA calendar from August to December 2026 is defined by pivotal regulatory decisions that address severe unmet clinical needs across a spectrum of diseases. Oncology and rare diseases dominate the agenda, with Nuvalent's targeted TKIs (zidesamtinib and neladalkib), Exelixis's combination immunotherapy (zanzalintinib), and Capricor's cellular therapy (deramiocel) facing critical PDUFA dates. Simultaneously, decisions regarding BioXcel's IGALMI for outpatient psychiatric agitation, Moderna's mRNA-1010 seasonal flu vaccine, and Novo Nordisk's metabolic juggernaut CagriSema could fundamentally alter treatment paradigms for millions of patients globally. While these target action dates provide a reliable chronological framework for investors and clinicians, they remain subject to routine FDA extensions, and the ultimate clinical impact of these therapies will depend heavily on real-world safety data, physician adoption, and ongoing market access negotiations.