Scientific analysis of Bryan Johnson's longevity protocol
Geroscience and the Paradigm of Biological Aging
The pursuit of human life extension has transitioned from theoretical biology into applied clinical pharmacology, an emerging field formally referred to as geroscience. The foundational hypothesis of geroscience posits that biological aging is not a rigid, chronological inevitability, but rather a malleable physiological process that serves as the primary risk factor for the majority of chronic diseases, including neurodegeneration, cardiovascular disease, and metabolic syndrome 1234. By targeting the underlying hallmarks of biological aging - such as cellular senescence, mitochondrial dysfunction, epigenetic alterations, and deregulated nutrient sensing - geroscience aims to delay the onset of multiple age-related morbidities simultaneously 14567. The ultimate clinical objective is the extension of "healthspan," defined as the period of life completely free from chronic, debilitating age-related disease 1567.
The Blueprint protocol, an intensive biological aging intervention program popularized by technology entrepreneur Bryan Johnson, operates directly on this geroscience premise. The protocol utilizes a highly quantified regimen of exhaustive biomarker testing, severe dietary restriction, and a polypharmacy stack of over 100 daily supplements and off-label prescription medications designed to optimize systemic physiology 891010.
A critical distinction within geroscience must be established when evaluating such protocols: the fundamental difference between extending healthspan and extending maximum lifespan. Standard medical interventions, and even some highly effective geroprotective interventions, reliably compress morbidity and increase median lifespan by preventing early mortality from specific diseases 5611. For example, the pharmacological clearance of senescent cells in murine models increases average median lifespan by approximately 25%, but it has virtually no effect on maximum lifespan; the longest-living treated subjects die at the same chronological age as the longest-living untreated control subjects 11. The aging vector that ultimately causes mortality is largely distinct from the vector that induces chronic illness 11. Caloric restriction remains one of the only interventions proven to significantly extend absolute maximum lifespan in mammalian models by fundamentally slowing the intrinsic rate of aging 611. This distinction between morbidity compression (healthspan) and age deceleration (lifespan) is vital for interpreting the efficacy, safety, and limitations of the individual pharmacological and nutraceutical components within the Blueprint protocol 1012.
Metabolic Modulators and Polypharmacy Interactions
A cornerstone of the Blueprint protocol involves the aggressive off-label use of metabolic drugs traditionally prescribed for the management of type 2 diabetes mellitus (T2DM). The combinatorial use of biguanides, alpha-glucosidase inhibitors, and sodium-glucose cotransporter-2 (SGLT2) inhibitors is engineered to suppress insulin spikes, artificially mimic a prolonged fasting state, and regulate critical nutrient-sensing pathways such as AMP-activated protein kinase (AMPK) and the mechanistic target of rapamycin (mTOR) 13151415. While individually efficacious for glycemic control, the convergence of these agents in normoglycemic individuals introduces complex pharmacokinetic and pharmacodynamic interactions.
Metformin and Acarbose
Metformin is an oral antihyperglycemic agent that reduces hepatic glucose production and enhances peripheral tissue insulin sensitivity, primarily functioning as a potent AMPK activator 151617. Acarbose, conversely, is an alpha-glucosidase inhibitor that acts directly within the gastrointestinal tract to decelerate the enzymatic breakdown and absorption of dietary carbohydrates, thereby effectively blunting postprandial glucose and insulin surges 181922. In preclinical murine models, particularly within the rigorously controlled National Institute on Aging's Interventions Testing Program (ITP), acarbose has consistently demonstrated robust lifespan extension, especially when administered as part of a combination therapy framework 23.
When coadministered, metformin and acarbose exhibit a highly distinct pharmacokinetic interaction. Acarbose alters overall gastrointestinal motility and specifically delays intestinal absorption rates, which has been shown to significantly reduce the systemic bioavailability of coadministered metformin 182420. Clinical pharmacokinetic studies consistently demonstrate that acarbose reduces both the peak serum concentration ($C_{max}$) and the area under the curve (AUC) of metformin by approximately 34% to 35% 2420. Despite this notable reduction in systemic metformin exposure, the pharmacodynamic effect on overall glycemic control remains highly synergistic and clinically beneficial. In extensive clinical trials, such as the MARCH (Metformin and Acarbose in Chinese as the initial Hypoglycemic treatment) trial, the fixed-dose combination of metformin and acarbose successfully reduced hemoglobin A1c (HbA1c), fasting blood glucose, and two-hour postprandial glucose without inducing a significant risk of hypoglycemia 142220212223.
However, long-term administration of this drug combination introduces highly specific systemic risks that require intensive monitoring. Metformin therapy is strongly and independently associated with vitamin B12 malabsorption, an effect that is severely exacerbated by prolonged, multi-year use 161724252627. Data drawn from the 13-year Diabetes Prevention Program Outcomes Study (DPPOS), which tracked 1,073 patients on continuous metformin versus 1,082 on placebo, revealed that accumulated years of metformin use increased the absolute risk of biochemical B12 deficiency by 13% for every single year of ongoing use (Odds Ratio 1.13; 95% CI, 1.06 - 1.20) 252627. This severe deficiency can manifest clinically as megaloblastic anemia, irreversible peripheral neuropathy, and elevated systemic homocysteine levels 1624252627. Consequently, the inclusion of long-term metformin in any longevity protocol necessitates vigilant annual testing of serum B12 and methylmalonic acid (MMA) alongside aggressive sublingual or injectable B12 supplementation 1624252627.
SGLT2 Inhibitors and the Risk of Euglycemic Ketoacidosis
SGLT2 inhibitors, representing a newer class of antihyperglycemic agents that includes dapagliflozin and empagliflozin, reduce blood glucose through an entirely distinct mechanism: they selectively inhibit glucose reabsorption in the proximal renal tubule, forcing the continuous excretion of glucose through the urine (glycosuria) 15171928. In recent years, SGLT2 inhibitors have emerged as frontline geroprotective candidates due to their profound cardiovascular and renal benefits. Large-scale trials and meta-analyses demonstrate that SGLT2 inhibitors significantly reduce heart failure hospitalizations, all-cause mortality, and the progression of chronic kidney disease, notably doing so completely independent of a patient's baseline diabetes status 2930313233.
Mechanistically, SGLT2 inhibitors promote a systemic metabolic shift toward ketogenesis, decrease harmful leptin release, and upregulate the longevity-associated sirtuin-1 protein, artificially mimicking a continuous fasting state that drastically reduces oxidative and endoplasmic reticulum stress 32. The efficacy of these mechanisms is supported by preclinical data from the ITP, where SGLT2 inhibitors successfully improved median longevity in male mice by 13.6% 23.
From a purely pharmacokinetic standpoint, SGLT2 inhibitors and metformin exhibit minimal overlap; metformin essentially does not bind to plasma proteins, whereas SGLT2 inhibitors are highly protein-bound (ranging from 86.2% to 99%), preventing direct competitive metabolic interactions 1528. However, a severe and potentially lethal pharmacodynamic risk emerges rapidly when SGLT2 inhibitors are combined with high-dose acarbose 19.
SGLT2 inhibitors routinely induce the forced renal excretion of 50 to 100 grams of glucose per day 19. Simultaneously, high-dose acarbose blocks the intestinal absorption of incoming dietary carbohydrates 19. The concurrent application of these two mechanisms mimics a state of extreme caloric starvation and severe carbohydrate depletion.
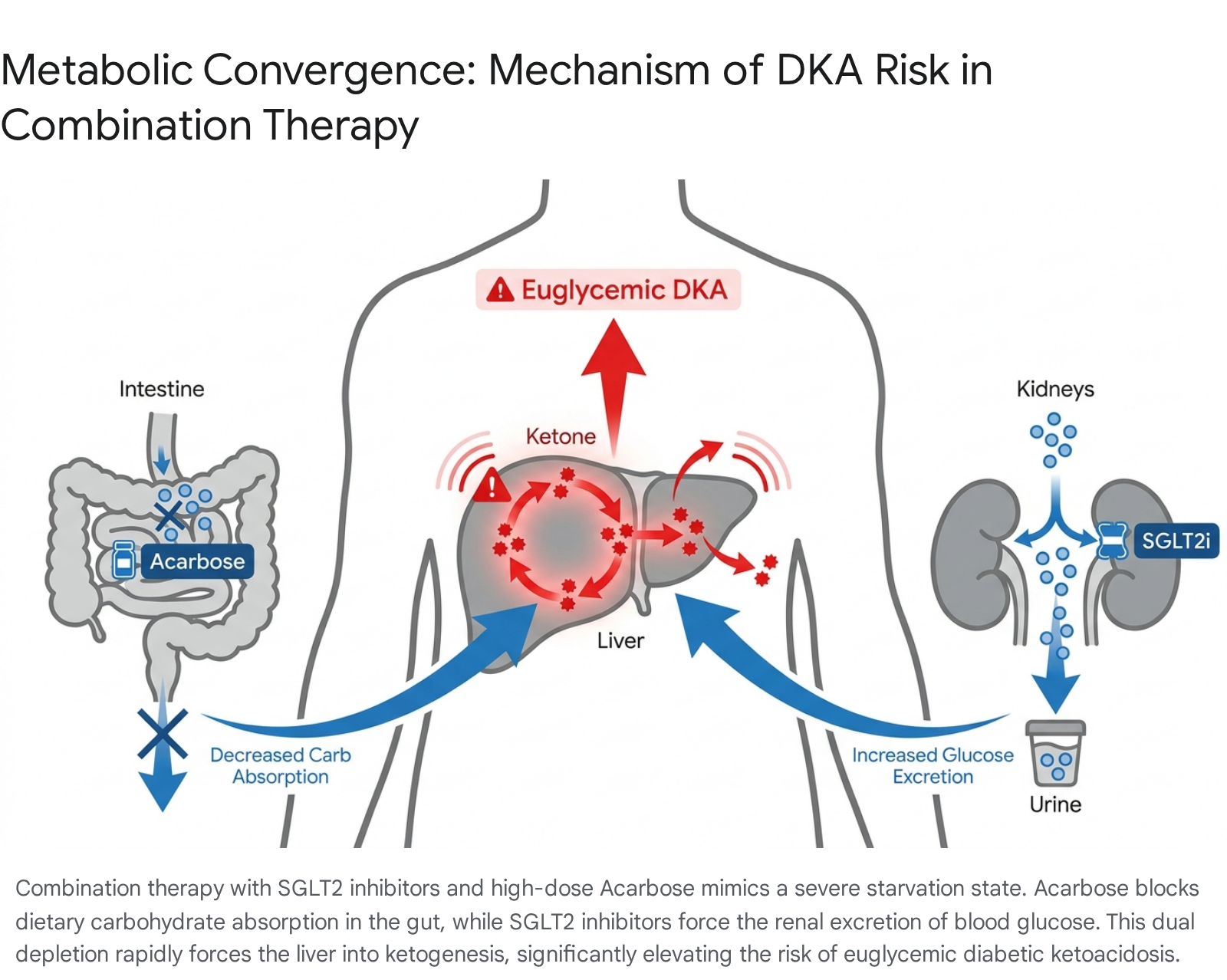
In individuals with compromised beta-cell function, or healthy individuals following strict low-carbohydrate and low-calorie diets, this specific combination drastically elevates the risk of euglycemic diabetic ketoacidosis (DKA) 151928.
Recent clinical case reports have documented healthy Asian patients treated with dapagliflozin (10 mg/day) and high-dose acarbose (300 mg/day) who rapidly developed severe diabetic ketosis without significant initial acidosis 19. Consequently, the prevailing medical consensus strongly advises extreme caution when attempting to combine SGLT2 inhibitors with high-dose alpha-glucosidase inhibitors, noting that the physiological toll of this combination in normoglycemic individuals can easily outweigh the theoretical geroprotective benefits 19.
| Drug Class | Example Agents | Primary Mechanism | Geroprotective / Longevity Evidence | Key Adverse Effects & Interactions |
|---|---|---|---|---|
| Biguanides | Metformin | AMPK activation; profound inhibition of hepatic gluconeogenesis 151528 | Modest lifespan extension in select animal models; widespread human epidemiological associations 101516. | Long-term use linearly increases the risk of Vitamin B12 deficiency (13% increased risk per year of ongoing use) 2527. |
| Alpha-Glucosidase Inhibitors | Acarbose | Delays intestinal carbohydrate breakdown and absorption 181922 | Significant maximum lifespan extension in mice (ITP), highly synergistic in combination 1523. | GI distress; drastically decreases systemic bioavailability of coadministered metformin by up to 35% 182421. |
| SGLT2 Inhibitors | Dapagliflozin, Empagliflozin | Inhibits renal glucose reabsorption, promoting continuous glycosuria 1517 | 13.6% median lifespan increase in male mice (ITP); globally proven human cardiorenal protection 233032. | Severe risk of euglycemic DKA when combined with high-dose acarbose or extreme low-calorie diets 1519. |
Discontinued Interventions: Rapamycin and Methylene Blue
The dynamic and highly quantified nature of intensive biological monitoring requires the immediate cessation of interventions when biomarker data indicates adverse systemic outcomes. Two prominent components of the historical Blueprint protocol - rapamycin and methylene blue - were deliberately discontinued following severely negative physiological feedback.
Rapamycin is a macrolide compound that acts as a specific inhibitor of mTOR, a central protein kinase that acts as the master regulator of cellular growth, proliferation, and survival 1039. Targeted inhibition of the mTORC1 complex effectively limits cellular growth signals and strongly upregulates autophagy, a cellular cleanup process essential for maintaining tissue homeostasis and clearing damaged organelles 1039. Historically, rapamycin remains the most rigorously validated pharmacological lifespan extender in mammalian models. Within the ITP, rapamycin administered at 600 days of age extended total mouse lifespan by up to 48% in females and 52% in males 1023. Notably, the combination of rapamycin and acarbose yielded an even greater median lifespan increase of 36.6% across diverse models 23.
However, translating the success of rapamycin to healthy, normoglycemic human populations introduces severe complications, primarily due to the drug's unavoidable concurrent inhibition of the mTORC2 complex during chronic or high-dose administration 39. Unlike mTORC1, mTORC2 is intimately tied to insulin signaling and systemic glucose homeostasis, largely mediated through the activation of kinases such as AKT and PKC 39. Excessive or chronic inhibition of mTORC2 rapidly leads to marked insulin resistance, dysregulated lipid profiles, and broad-spectrum immunosuppression 393435.
After nearly five years of testing various rapamycin dosing protocols (including weekly 5, 6, and 10 mg schedules, as well as biweekly 13 mg pulses), Johnson definitively discontinued the drug in September 2024 39343536. The cessation was directly prompted by persistent and accumulating side effects, including intermittent skin and soft tissue infections (a hallmark of clinical immunosuppression and inhibited natural killer cells), severe lipid abnormalities, steadily elevated blood glucose, and a noticeably increased resting heart rate 34353637. Furthermore, new pre-print epigenetic data evaluated in late 2024 suggested that continuous rapamycin therapy might paradoxically accelerate epigenetic aging markers across 16 different biological clocks in humans, prompting its immediate removal from the protocol to prioritize actual healthspan over theoretical longevity 393537.
Similarly, methylene blue, a synthetic compound historically utilized in biohacking circles for its unique redox properties and alleged ability to act as an alternative electron carrier within the mitochondrial electron transport chain, was abruptly abandoned. The intervention was terminated after just 13 days of use in August 2025 due to a direct, negative pharmacological interaction with intermittent hypoxia therapy; the compound actively interfered with the physiological efficacy of the oxygen treatments and induced severe neurological headaches, starkly demonstrating the unforeseen pharmacological conflicts inherent in extreme, multi-agent polypharmacy 34.
Cellular Energetics and Epigenetic Modulators
Beyond prescription medications, longevity protocols rely heavily on high doses of natural compounds, metabolic precursors, and synthesized metabolites aimed at restoring fundamental cellular energy pathways, reducing oxidative stress, and maintaining genomic and epigenetic stability.
NAD+ Precursors: Nicotinamide Riboside (NR) vs. Nicotinamide Mononucleotide (NMN)
Nicotinamide adenine dinucleotide (NAD+) is an indispensable coenzyme present in all living cells, essential for fundamental energy metabolism, robust mitochondrial function, and the structural activation of sirtuins (a highly conserved family of longevity-linked proteins) 154438. Clinical data reveals that plasma NAD+ concentrations decline by approximately 60% over the human lifespan, dropping sharply from around 50 nanomolar in healthy young adults (ages 20 - 40) to a mere 20 nanomolar in the elderly (ages 60 - 97), severely impairing DNA repair mechanisms, stem cell activity, and overall cellular resilience 1546.
To theoretically restore youthful NAD+ levels, modern longevity protocols heavily utilize NAD+ precursors, primarily focusing on nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN). Both of these compounds are clinically proven to safely elevate circulating NAD+ in humans at standard doses ranging from 250 mg to 1,000 mg per day without major adverse events 443846. However, the relative efficacy and cellular absorption mechanisms of these two specific precursors have been heavily debated within the geroscience community. Biologically, NMN must be enzymatically converted into NR in the extracellular space before it can enter most cell types; conversely, NR enters cells directly via specific nucleoside transporters, granting it a distinct structural and metabolic efficiency advantage 3847.
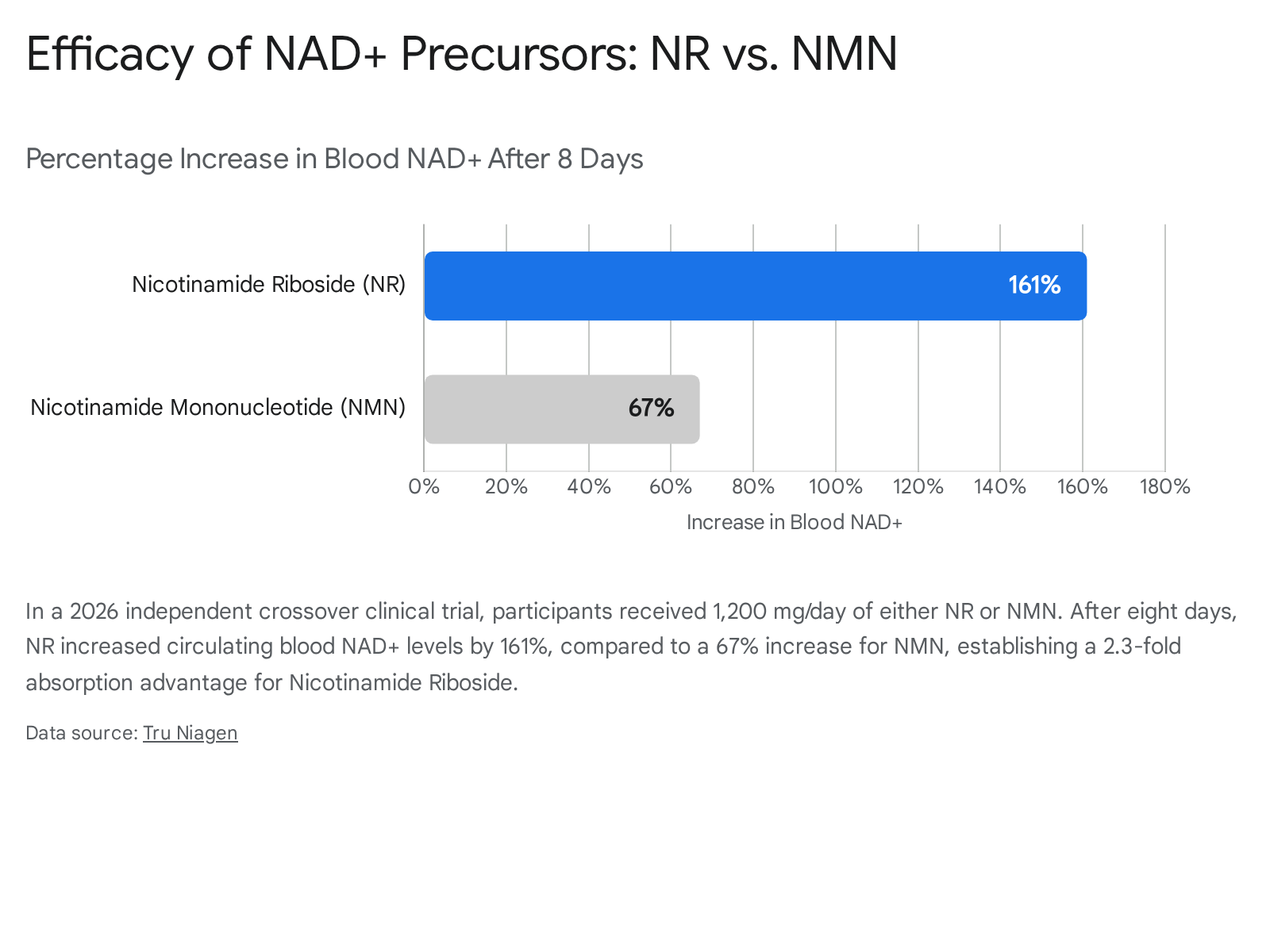
This specific mechanistic advantage was corroborated in early 2026 by an independent, peer-reviewed crossover human clinical trial published in the Cell Press journal iScience 47. In a direct, placebo-controlled head-to-head comparison utilizing a high dosage of 1,200 mg/day, NR successfully elevated blood NAD+ levels by an impressive 161% after eight days of supplementation. In the exact same patient cohort, an equivalent 1,200 mg/day dose of NMN elevated NAD+ by only 67% - demonstrating a highly significant 2.3-fold pharmacokinetic advantage for Nicotinamide Riboside 47.
An alternate 2026 study published in Nature Metabolism indicated that after an extended 14 days of supplementation, both NR and NMN effectively doubled circulating NAD+ levels compared to placebo, but it uniquely revealed that both compounds are heavily processed and altered by the gut microbiome into nicotinic acid (NA) prior to systemic absorption, complicating the direct tissue-delivery models previously assumed 48. Furthermore, despite these robust, measured increases in blood NAD+, long-term human clinical trials have yet to demonstrate profound physiological or functional gains (e.g., the reversal of cognitive decline, significant increases in walking speed, or vast cardiovascular improvements) over short durations, indicating that simply elevating blood NAD+ may not uniformly translate to measurable improvements in tissue-level healthspan 46.
Calcium Alpha-Ketoglutarate (CaAKG)
Alpha-ketoglutarate (AKG) is a core endogenous metabolic intermediate deeply embedded within the mitochondrial tricarboxylic acid (TCA) cycle, playing a fundamentally pivotal role in cellular energy metabolism, amino acid synthesis, and the continuous maintenance of redox balance 395051. More importantly for the field of geroscience, AKG serves as an absolutely mandatory substrate and co-factor for TET (Ten-Eleven Translocation) and Jumonji C-domain-containing enzymes, which facilitate the specific demethylation of DNA and histones 13. By structurally regulating these epigenetic enzymes, AKG possesses the theoretical biological capacity to actively reverse age-related epigenetic shifts in gene expression. In extensive mammalian models, late-life supplementation with AKG significantly reduced systemic frailty, heavily enhanced stress resistance, and modestly increased overall lifespan (by approximately 4%) 5051.
Because free un-bound AKG demonstrates exceptionally poor oral bioavailability, low chemical stability, and rapid degradation in the gut, advanced longevity protocols exclusively utilize its stabilized salt form, Calcium Alpha-Ketoglutarate (CaAKG) 395040. High-quality human clinical evidence for CaAKG has begun emerging rapidly. Initial retrospective observational data tracking mid-life adults taking 1 gram of CaAKG daily for seven months indicated a striking average reduction in DNA methylation biological age of up to 8 years, without any serious adverse effects reported 13.
In 2025 and 2026, scientific research into CaAKG expanded significantly into the realm of neuroprotection. Groundbreaking preclinical data published in Aging Cell (2025) by researchers at the National University of Singapore demonstrated that CaAKG completely restores synaptic plasticity and associative memory in severe Alzheimer's disease models 4142. It achieves this by aggressively upregulating neuronal autophagy to clear damaged proteins and re-establishing long-term potentiation (LTP) - the core neurological process that allows neurons to structurally strengthen their connections to form memories 4142. To definitively validate human efficacy, multiple large-scale randomized placebo-controlled trials are actively underway as of 2026, including a rigorous 12-week trial specifically evaluating CaAKG's impact on biological aging (utilizing the PhenoAge clock), glucose metabolism, and systemic inflammatory markers in adults aged 45 to 75 394043. At present, human data confirms that sustained doses of up to 4.5 to 6 grams per day are completely safe, highly well-tolerated, and yield modest but measurable improvements in bone mineral density and nitrogen balance (a marker of muscle maintenance) in the elderly 135156.
Senolytics and Autophagy Inducers
A central pillar of the geroscience framework is the active clearance of cellular damage that naturally accumulates as a byproduct of metabolism. This clearance is pursued through two primary modalities: inducing apoptosis in damaged, senescent cells (senolytics) and upregulating the internal cellular recycling machinery (autophagy).
Fisetin: Senolytic Targeting of the SASP
Cellular senescence is a protective state of irreversible cell cycle arrest triggered in response to severe genomic or oxidative stress, preventing damaged cells from proliferating 4445. While initially serving as a crucial tumor suppressor mechanism, the gradual, un-cleared accumulation of senescent cells directly drives systemic tissue dysfunction through the continuous, toxic secretion of pro-inflammatory cytokines, proteases, and chemokines - a phenomenon collectively known as the senescence-associated secretory phenotype (SASP) 4445.
Fisetin, a naturally occurring flavonoid polyphenol found in varying concentrations in strawberries, apples, and onions, has been identified through rigorous preclinical screening as one of the most potent natural "senolytics" - compounds capable of selectively inducing apoptosis only in senescent cells while sparing healthy tissue 444546. In aged wild-type mice, acute or intermittent administration of fisetin dramatically reduced the absolute burden of senescent cells in adipose and human explant tissue, dampened widespread SASP inflammatory markers (including IL-6 and IL-8), and successfully extended both median and maximum lifespan, restoring overall tissue homeostasis 444546.
Despite these profound preclinical results, the human translation of fisetin is currently stalled in the preliminary pilot phase. A 2024 pilot study meticulously investigated a heavy dosage protocol of 500 mg of fisetin administered for one week per month over a six-month period in healthy human adults 47. The physiological results were notably mixed and somewhat concerning: while four out of ten participants experienced a measurable reduction in biological age, five out of ten participants actually experienced an increase in their biological age, and telomere lengths remained entirely unchanged across the board 47. No overt adverse effects were reported among the subjects, but researchers unequivocally concluded that standalone fisetin supplementation cannot be recommended as a reliable anti-aging agent in healthy humans until results from vastly larger, placebo-controlled trials - such as the ongoing AFFIRM-LITE trial evaluating frailty, insulin resistance, and inflammation in adults aged 70 to 90 - are published and peer-reviewed 464748.
Spermidine and Polyamine Homeostasis
Spermidine, a natural polyamine deeply involved in fundamental intermediate metabolism, functions as a highly potent inducer of autophagy - the internal cellular maintenance process inherently responsible for degrading and recycling damaged organelles, misfolded proteins, and metabolic waste 4950. Spermidine achieves this systemic cleanup by directly acting as a biochemical inhibitor of EP300 (E1A-associated protein p300), a specific acetyltransferase that operates as an endogenous, negative inhibitor of autophagy 4950. By blocking EP300, spermidine allows the autophagy-regulatory circuitries to function unimpeded. In aging murine models, continuous spermidine supplementation successfully suppresses age-related cardiovascular decline and extends overall median longevity by approximately 10% 495051.
Uniquely, epidemiological evidence in humans robustly and independently correlates dietary spermidine intake with significantly reduced mortality. A comprehensive 20-year prospective cohort study demonstrated that individuals situated in the top third of dietary spermidine intake enjoyed a profound reduction in all-cause mortality, an effect mathematically equivalent to a biological age reduction of 5.7 years, completely independent of other caloric, socioeconomic, and lifestyle factors 505253.
However, translating this dietary epidemiology into isolated, high-dose pill supplementation presents severe pharmacokinetic challenges. A rigorous double-blind, randomized controlled trial published in late 2024 heavily investigated the clinical safety and pharmacokinetics of high-purity spermidine trihydrochloride (hpSPD) administered at 40 mg/day over 28 days in healthy older men 54. While the intervention was proven to be entirely safe and well-tolerated without any clinical adverse events, it resulted in only minimal, statistically insignificant changes to circulating polyamine concentrations in both the serum and urine 5154. Furthermore, meticulous 2023 pharmacokinetic data revealed that while short-term oral spermidine (15 mg/day) successfully increased plasma levels of spermine (a downstream polyamine metabolite), it completely failed to significantly elevate systemic levels of spermidine itself, highly likely due to rapid enzymatic metabolic conversion in the gut and extremely tight systemic homeostatic control mechanisms 55. Consequently, while its safety profile is excellent, the systemic bioavailability and cellular penetration of encapsulated oral spermidine in humans remains highly constrained, limiting its immediate utility as a pharmaceutical-grade geroprotector.
Neuroprotection and Trace Minerals
The Blueprint protocol frequently iterates on highly experimental compounds with exceptionally narrow therapeutic windows in the aggressive pursuit of neuroprotection, cognitive preservation, and molecular resilience against neurological decay.
The Toxicity and Failure of NDGA
Nordihydroguaiaretic acid (NDGA) is a potent antioxidant and anti-inflammatory phenolic compound historically derived from the leaves of the creosote bush 56. It operates physiologically by modulating the Nrf2 antioxidant pathway and directly inhibiting the p300 histone acetyltransferase, a key epigenetic regulator of autophagy 57. In preclinical testing within the ITP, NDGA yielded a 12% increase in median lifespan in male mice, sparking intense interest in its geroprotective potential; however, it yielded absolutely no maximum lifespan extension, and completely failed to provide any survival benefit to female mice 5658.
More concerningly for human application, NDGA exhibits severe, dose-dependent, and frequently fatal toxicity. Even at the exceptionally low doses explicitly utilized for the aforementioned lifespan studies in mice, NDGA significantly increased the incidence of severe liver, lung, and thymus tumors, and actively induced peritoneal hemorrhagic diathesis 5658. In humans, chronic high-dose exposure to NDGA is clinically associated with severe nephrotoxicity, fulminant liver failure, and the development of renal cell carcinoma 5658. Due to these extreme hazards, it was formally removed from the FDA's GRAS (Generally Recognized As Safe) list decades ago 5658. Consequently, the scientific rationale for NDGA inclusion in any healthy human longevity protocol is entirely unsupported by safety data, representing an unacceptable risk-to-reward ratio.
Low-Dose Lithium and Cognitive Preservation
Conversely, the implementation of trace-dose or low-dose lithium (frequently administered in the highly bioavailable form of lithium orotate) is gaining substantial, evidence-based traction within the geroscience community 72. While high-dose lithium carbonate (often exceeding 900 mg/day) remains the psychiatric standard for managing bipolar disorder, epidemiological studies consistently show that general populations exposed to trace lithium in their local drinking water (ranging from 0.1 to 0.3 mg/day) exhibit significantly lower all-cause mortality, reduced cardiovascular mortality, and a dramatically decreased incidence of Alzheimer's disease 59606176.
Mechanistically, lithium directly and potently inhibits glycogen synthase kinase-3 (GSK-3), a critical enzyme whose pathological overactivity is heavily linked to systemic inflammation, circadian rhythm disruption, and advanced neurodegeneration 596176. Furthermore, lithium promotes protective Wnt signaling pathways and increases the synthesis of brain-derived neurotrophic factor (BDNF), greatly enhancing neuronal resilience against stress 5961.
In late 2025, highly publicized research published in Nature indicated that localized lithium deficiency within the brain tissue itself may be a fundamental, early pathological driver of mild cognitive impairment (MCI) and subsequent Alzheimer's disease 60. Following this, a 2026 pilot randomized clinical trial (published in JAMA Neurology) meticulously investigated the use of low-dose lithium carbonate (approximately 195 mg/day) in older adults specifically diagnosed with MCI 6077. While the trial technically missed statistical significance on its six highly stringent primary endpoints (including total brain volume and composite cognition scores), it revealed a highly promising clinical trend: verbal memory declined at roughly half the rate in the lithium-treated group compared directly to the placebo cohort (P = .05) 6077. The safety profile of this low-dose regimen was excellent, with no serious adverse events linked to the medication 77. Researchers now suggest that future clinical trials utilizing the highly bioavailable lithium orotate formulation at micro-doses may yield much more definitive neuroprotective outcomes, establishing it as a primary candidate for cognitive longevity 7260.
Quantifying Efficacy: Biological Aging Clocks
To accurately quantify the physiological efficacy of these extreme interventions, longevity protocols inherently rely on advanced biological aging biomarkers. Conventional clinical markers (e.g., standard lipid panels, basic metabolic panels, HbA1c) are vital for disease monitoring but completely lack the sensitivity required to capture the cellular velocity of aging 3. Over the past decade, DNA methylation (DNAm) epigenetic clocks have become the definitive gold standard for estimating actual biological age .
However, first-generation clocks (which were trained simply to predict chronological age based on methylation patterns) have been entirely superseded by much more accurate, phenotypically anchored models. The current scientific consensus highlights two premier clocks: GrimAge (a second-generation clock heavily trained on mortality risk and health-related phenotypes) and DunedinPACE (a third-generation clock uniquely designed to track the longitudinal, real-time pace of biological aging) 62. In a highly comprehensive 2026 clinical evaluation of 14 different advanced aging biomarkers (spanning functional, inflammatory, and epigenetic measures), DunedinPACE emerged unequivocally as the single strongest and most consistent predictor of both all-cause and cause-specific mortality 63. Furthermore, unlike earlier static clocks, DunedinPACE is highly sensitive to short-term geroprotective interventions; data drawn from the landmark CALERIE trial definitively demonstrated that DunedinPACE could accurately and rapidly detect a significant slowing in the biological pace of aging in humans undergoing controlled caloric restriction, verifying its utility as a rapid-feedback metric for protocols like Blueprint 626482.
Additionally, 2025 saw the validation and deployment of highly precise, organ-specific proteomic aging clocks 836585. Utilizing high-plex analytical platforms (such as SomaScan) to evaluate thousands of individual plasma proteins, researchers demonstrated that individual organs (e.g., the brain, heart, kidneys) age on completely distinct, weakly correlated biological trajectories within the exact same individual 836586. Strikingly, nearly 1 in 5 ostensibly healthy adults possess at least one organ exhibiting severely accelerated aging (defined clinically as an organ age gap one full standard deviation higher than same-aged peers) 83. Brain aging is particularly critical in this context; an accelerated brain proteomic clock strongly predicts the future onset of Alzheimer's disease and is the single most potent predictor of overall systemic mortality, acting essentially as a central biological lifespan regulator 658666. The integration of the DunedinPACE algorithm alongside organ-specific proteomic profiling provides a rigorous, biologically interpretable framework to evaluate whether experimental protocols like Blueprint successfully retard systemic biological decay or merely alter superficial biomarkers 6465.
Conclusion
The pharmacological and nutraceutical interventions comprising the Blueprint protocol represent the extreme, highly experimental frontier of applied geroscience. An intensive analysis of the current scientific literature confirms that many foundational elements of the protocol - such as caloric restriction, NAD+ precursors, SGLT2 inhibitors, and targeted autophagy inducers like CaAKG - possess robust mechanistic backing and proven clinical benefits regarding morbidity compression and cardiovascular health.
However, the protocol's heavy reliance on intensive polypharmacy introduces profound, compounding physiological risks that frequently eclipse their theoretical longevity benefits. The severe euglycemic ketoacidosis risk presented by combining SGLT2 inhibitors with carbohydrate-blocking agents, the long-term immunosuppression and metabolic dysregulation associated with chronic rapamycin use, and the overt cellular toxicity of agents like NDGA highlight the extreme dangers of translating isolated animal lifespan data directly to human biological systems. The recent cessation of core therapeutics like rapamycin and methylene blue underscores a critical reality within geroscience: aggressive, multi-pathway pharmacological suppression of aging pathways often triggers unpredictable compensatory homeostatic disruptions. Ultimately, while interventions targeting epigenetic regulation and metabolic resilience show immense promise for significantly extending human healthspan, the reliable extension of maximum human lifespan through unguided, extreme polypharmacy remains biologically unproven.