Role of AMPK in metformin and exercise longevity
Introduction: The Metabolic Checkpoint of Aging and Geroscience
Aging is fundamentally characterized by a progressive, systemic loss of metabolic flexibility, leading to the gradual deterioration of tissue homeostasis and an exponentially increased vulnerability to chronic, non-communicable diseases. At the molecular center of this metabolic decline is the 5′ AMP-activated protein kinase (AMPK), a highly conserved serine/threonine kinase that functions as the master cellular energy sensor across nearly all eukaryotic cells 12. Functioning as a heterotrimeric complex comprising a catalytic $\alpha$ subunit alongside regulatory $\beta$ and $\gamma$ subunits, AMPK is acutely activated in response to energy depletion - specifically, an increase in the intracellular AMP-to-ATP or ADP-to-ATP ratios, signaling an energetic deficit 13. Once activated, primarily through the phosphorylation of the Thr172 residue on the $\alpha$ subunit by upstream kinases such as Liver Kinase B1 (LKB1), AMPK initiates a profound, systemic metabolic rewiring 134. It suppresses energy-consuming anabolic pathways, such as protein and lipid synthesis, primarily via the direct inhibition of the mechanistic target of rapamycin complex 1 (mTORC1), while simultaneously upregulating energy-generating catabolic processes, including glucose uptake, fatty acid $\beta$-oxidation, and macroautophagy 1345.
Because the biological process of aging is driven in large part by the gradual accumulation of oxidative damage, mitochondrial dysfunction, defective proteostasis, and the aberrant hyperactivation of nutrient-sensing pathways like mTOR, AMPK has emerged as a premier target for gerotherapeutic interventions 467. For decades, the ubiquitous type 2 diabetes mellitus (T2DM) medication metformin, alongside structured physical exercise, has been championed as the most accessible and effective method to stimulate AMPK and theoretically delay biological aging 36. Both interventions independently improve whole-body insulin sensitivity, enhance cardiovascular function, and trigger cellular quality control mechanisms 68. This perceived synergistic potential led to widespread, deeply entrenched clinical guidelines recommending the automatic co-prescription of metformin alongside rigorous exercise regimens for patients presenting with metabolic syndrome, prediabetes, or overt T2DM 91011.
However, a surge of human clinical data, advanced multi-omics analyses, and exhaustive systematic reviews published between 2023 and 2026 have fundamentally challenged the long-held assumption that combining these two interventions yields additive benefits 9101213. Recent investigations unequivocally demonstrate that combining metformin with aerobic or progressive resistance training frequently results in a stark antagonistic interaction, whereby the pharmacological agent blunts the critical cardiovascular, mitochondrial, and transcriptional adaptations normally induced by physical exertion 13141516. Furthermore, an advanced understanding of AMPK's tissue-specific isoforms has highlighted the "Goldilocks effect" - the physiological reality that chronic, non-specific AMPK activation carries significant maladaptive risks, including pathological cardiac hypertrophy and enhanced survival capacities for established tumor cells 21717.
This comprehensive report dissects the rapidly evolving landscape of AMPK signaling, prioritizing 2023 - 2026 human clinical trials, epidemiological updates, and systematic reviews. It scrutinizes the precise molecular mechanisms of metformin-exercise antagonism, evaluates the longevity paradox of metformin in healthy versus diabetic populations (including the stalled status of the Targeting Aging with Metformin [TAME] trial), explores the severe risks of chronic AMPK activation, and analyzes the paradigm shift toward tissue-specific, isoform-selective next-generation AMPK activators across diverse demographic populations.
The Metformin-Exercise Antagonism: Deconstructing the Blunting Effect
For many years, the standard clinical practice of prescribing metformin alongside structured physical activity was predicated on the premise that both modalities act upon overlapping, yet distinct, metabolic pathways to cumulatively improve glycemic control and cardiovascular fitness. While this holds true for baseline glycemic management in individuals with advanced, overt T2DM, extensive clinical evidence now confirms a pervasive and highly significant blunting effect when the two interventions are combined, particularly concerning physical adaptation, aerobic capacity, and vascular health 131619.
Evidence from 2024-2026 Systematic Reviews and Meta-Analyses
The paradigm shift regarding combined therapy was solidified by several comprehensive systematic reviews and network meta-analyses published between 2024 and 2026 13161918. A pivotal 2026 meta-analysis encompassing nine rigorous human trials (n=827) systematically quantified the interaction between metformin and exercise across the entire spectrum of glucose dysregulation. The aggregated data revealed that individuals receiving metformin in conjunction with exercise experienced significantly smaller improvements in peak oxygen uptake (VO2peak) compared to those undertaking the exact same exercise regimen alongside a placebo (Mean Difference [MD] - 1.19 mL/kg/min; 95% CI - 2.33 to - 0.04) 13.
Furthermore, metformin significantly attenuated the exercise-induced reductions in both systolic blood pressure (an attenuation of 3.76 mmHg) and diastolic blood pressure (an attenuation of 1.98 mmHg) 13. These physiological dampening effects were observed regardless of the specific exercise intensity applied, complicating earlier clinical assumptions that high-intensity interval training might override pharmacological blunting 91215. Participants taking metformin also reported higher rates of perceived exertion during exercise and lower overall reductions in resting heart rate, suggesting that the drug alters the systemic physiological response to physical stress without conferring additional performance or fitness advantages 1321.
Demographic Divergence: Prediabetes vs. Type 2 Diabetes
A corollary network meta-analysis comprising 407 articles and over 33,802 participants parsed these interactions by demographic and disease state, identifying a crucial divergence in therapeutic efficacy between prediabetic and T2DM populations 18. In patients with advanced T2DM, metformin monotherapy proved significantly more efficacious than exercise alone in reducing HbA1c ( - 0.88% vs. - 0.48%), 2-hour glucose levels, and fasting glucose 1918. This indicates that when metabolic dysfunction is severe and intrinsic metabolic flexibility is lost, pharmacological intervention remains superior for immediate glycemic control.
However, in prediabetic individuals, the hierarchy of efficacy inverted entirely. Exercise unequivocally outperformed metformin monotherapy in reducing HbA1c ( - 0.16% vs. - 0.10%), 2-hour glucose ( - 0.68 mmol/L vs. 0.01 mmol/L), and Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) 1918. Crucially, when prediabetic individuals combined the therapies, the addition of metformin severely restricted the maximum potential gains. This phenomenon highlights a vital demographic nuance: the utility of metformin is inversely proportional to the baseline metabolic health of the patient. In a relatively healthier prediabetic population, the introduction of a systemic metabolic brake like metformin interferes with the body's natural capacity to adapt to physiological stressors, effectively capping the benefits of lifestyle interventions 10111819.
The Rutgers Human Clinical Trial on Vascular Insulin Sensitivity
The macroscopic epidemiological data was corroborated by precise physiological measurements in a landmark 2025 double-blind, placebo-controlled trial conducted by researchers at Rutgers University 121519. The study enrolled 72 adult participants who were at high risk for metabolic syndrome - a dangerous clustering of obesity, hypertension, and abnormal cholesterol - and separated them into four highly controlled groups: high-intensity exercise with placebo, high-intensity exercise with metformin, low-intensity exercise with placebo, and low-intensity exercise with metformin 1011. Over a 16-week period, researchers tracked alterations in vascular insulin sensitivity, a critical metric defining how well blood vessels dilate to shuttle glucose and oxygen out of the bloodstream and into skeletal muscle tissues following a meal 1019.
The results were unequivocal: exercise alone drastically improved macrovascular and microvascular insulin sensitivity, enhancing capillary blood flow and expanding conduit arteries 101119. However, the introduction of 2 grams of daily metformin blunted these exercise-training-mediated increases in vascular insulin sensitivity at the level of both conduit arteries and capillaries, operating in parallel with diminished aerobic fitness gains and altered inflammatory profiles 1219. The blunting effect occurred irrespective of the exercise intensity, confirming that metformin inherently dampens the vascular adaptations required for long-term cardiovascular health in non-diabetic populations 101215.
Molecular Mechanisms of Antagonism: ROS Suppression and Transcriptional Repression
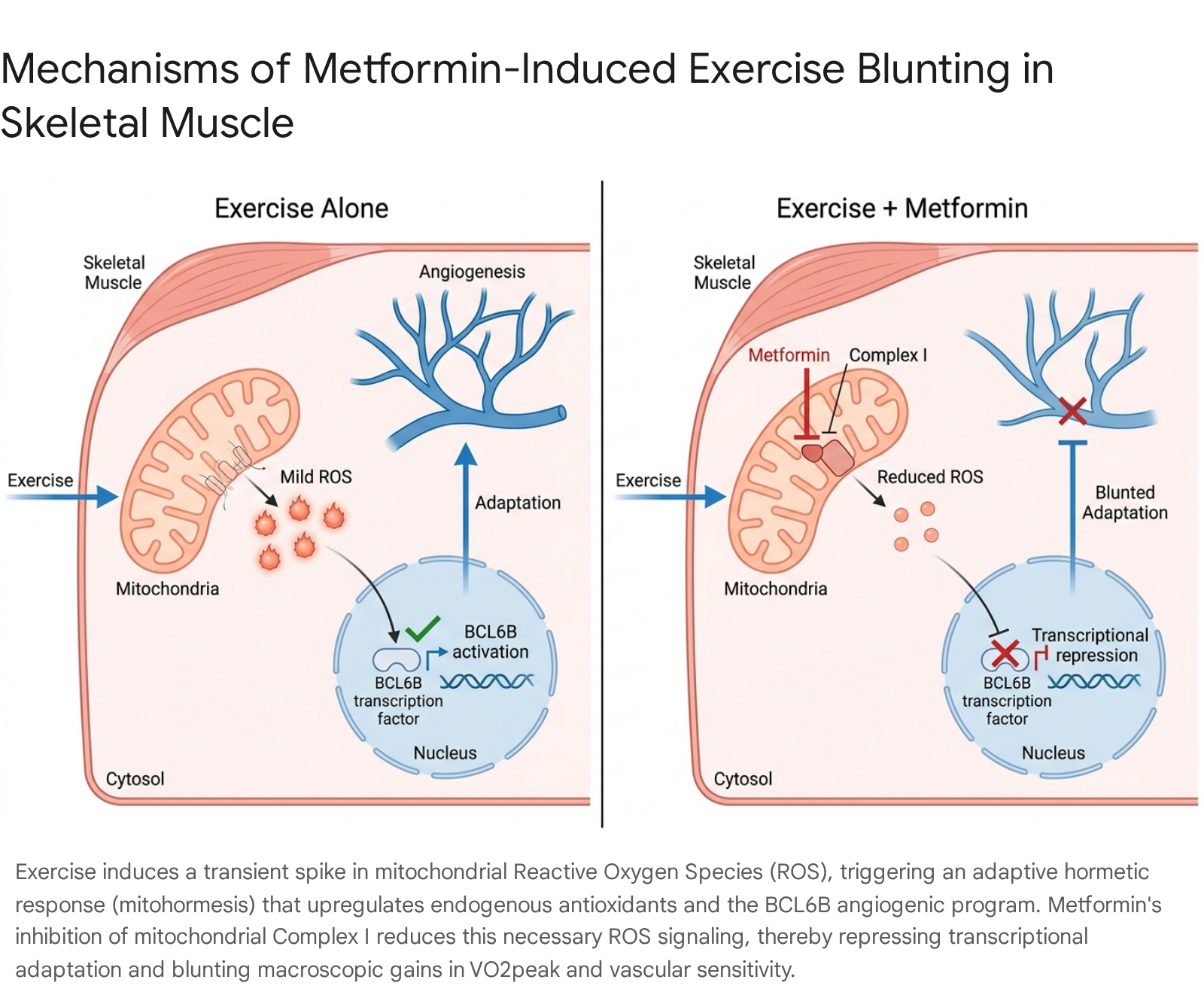
To fully comprehend why a widely prescribed geroprotector undermines the most potent physiological longevity intervention known to human medicine (exercise), researchers have turned to the sub-cellular dynamics of mitochondrial respiration and transcriptional signaling. The prevailing explanation for this profound interference is rooted in the concept of mitohormesis and the suppression of reactive oxygen species (ROS), alongside downstream epigenetic repression 3202421.
The Mitohormesis Hypothesis and ROS Quenching
Metformin primarily exerts its cellular effects by accumulating within the mitochondrial matrix, where it acts as a mild, reversible inhibitor of the electron transport chain at Complex I 3915. This deliberate inhibition lowers the efficiency of cellular ATP production, leading to an energy deficit (a high AMP/ATP ratio) that subsequently activates AMPK. Consequently, AMPK activation suppresses mTORC1, halting anabolic cell growth in favor of cellular repair and autophagy 34.
However, a highly consequential secondary effect of Complex I inhibition by metformin is a profound reduction in the generation of mitochondrial reactive oxygen species (mtROS) 392024. Historically, ROS were viewed almost exclusively as toxic, destructive byproducts of cellular metabolism that drove the aging process via cumulative oxidative damage 420. Modern geroscience has heavily refined this outdated view through the lens of mitohormesis - the biological principle that mild, transient bursts of oxidative stress serve as vital, indispensable intracellular signaling molecules 62024.
During strenuous physical exercise, skeletal muscle mitochondria generate a highly controlled, transient burst of ROS. This acute oxidative spike acts as a critical messenger, activating stress-response kinases, shifting the SIRT1-GSK3$\beta$ signaling axis, and signaling the nucleus to drastically upregulate endogenous antioxidant defense systems, enhance mitochondrial biogenesis, and improve vascular insulin sensitivity 2024. By chemically suppressing this exercise-induced ROS spike at its source in Complex I, metformin effectively "silences" the cellular alarm bell. Without the ROS signal, the skeletal muscle fails to recognize the true magnitude of the physiological stress it just endured, resulting in an aborted adaptive response 910. The absence of this acute stress signal directly prevents the expansion of mitochondrial density and aerobic capacity, providing a clear mechanistic explanation for why individuals on metformin exhibit stunted improvements in VO2peak despite rigorous training regimens 101215.
Transcriptional Repression and the BCL6B Angiogenic Program
Moving beyond acute ROS suppression, the metformin-exercise antagonism is deeply entrenched in the realm of transcriptional repression. Groundbreaking transcriptomic research published in late 2025 by investigators at Rutgers University and the University of Wisconsin-Madison illuminated these specific downstream mechanisms by performing parallel analyses of muscle biopsies from human subjects and murine models 212223.
In a highly controlled study investigating the effects of progressive aerobic exercise training (AET) combined with metformin in mice, comprehensive RNA sequencing of skeletal muscle revealed that metformin decreased the total number of exercise-induced differentially expressed genes by approximately 50% compared to mice undergoing exercise alone 2123. The addition of metformin broadly and indiscriminately suppressed multiple vital transcription factors and complex signal transduction pathways essential for skeletal muscle proteostasis, myogenesis, and overall oxidative capacity 2124.
Crucially, a parallel multi-omics analysis of human resistance exercise data uncovered a conserved, metformin-sensitive regulatory axis centered on the transcription factor B-Cell CLL/Lymphoma 6B (BCL6B) 142223. BCL6B, which is highly enriched in endothelial cells and adipocytes across both humans and murine models, serves as a primary driver of exercise-induced angiogenesis - the critical formation of new capillary networks required to supply oxygen and nutrients to actively hypertrophying muscle fibers 14212223. Metformin administration was shown to profoundly repress this specific BCL6B-associated signaling network 142324. By stunting angiogenesis at the foundational level of gene expression, metformin essentially starves the muscle tissue of the microvascular adaptations required for enhanced insulin sensitivity and cardiovascular fitness 101219. This molecular evidence perfectly aligns with the macroscopic clinical observations of blunted vascular dilation, restricted blood flow, and impaired muscle hypertrophy in adults taking the drug during progressive resistance training 111421.
Scrutinizing Metformin's Longevity Benefits: Healthy vs. Diabetic Populations
If metformin inherently blunts physiological adaptation and blunts angiogenesis, does it still possess the profound anti-aging properties that catapulted it to the forefront of longevity medicine? The clinical answer appears to be highly context-dependent, revealing a stark dichotomy between its biological effects in pathologically compromised individuals versus healthy, disease-free populations.
Epidemiological Origins and the 20-Year Survival Reversal
The modern enthusiasm for metformin as an anti-aging therapeutic originated largely from a landmark 2014 observational study published in the UK, which encompassed over 180,000 patients. The study revealed a striking, counterintuitive paradox: patients with T2DM treated with metformin monotherapy exhibited a slightly lower all-cause mortality rate than age-matched, non-diabetic healthy controls 331. Given that a diagnosis of T2DM typically truncates an individual's lifespan by 8 to 10 years, the observation that a diseased cohort outlived a healthy cohort catalyzed the hypothesis that metformin was actively slowing the fundamental biological aging process independent of its glycemic effects 331.
However, subsequent and vastly more rigorous long-term longitudinal analyses have significantly tempered this early enthusiasm. A recent, comprehensive 2025 review utilizing the Secure Anonymised Information Linkage dataset in Wales tracked T2DM patients treated with metformin (N=129,140) versus those treated with sulfonylureas (N=68,563), matched against healthy non-diabetic controls over a simulated 20-year period 325. The researchers controlled tightly for diverse demographic factors including age, sex, smoking status, and prior history of cancer and cardiovascular disease.
The findings painted a much more complex picture of longevity. While metformin users maintained a clear, undeniable survival advantage over patients prescribed sulfonylureas, the initial longevity benefit observed relative to matched healthy controls eroded over the extended timeline 325. Within the first three to five years of treatment, metformin did indeed confer a protective survival benefit over non-diabetics; however, when the observation period extended toward twenty years, the cumulative systemic toll of the underlying T2DM pathology eventually outweighed the drug's protective benefits. Ultimately, the long-term metformin cohort exhibited a shorter overall survival time than the healthy control group 25.
This longitudinal reversal underscores a vital pharmacological reality: metformin is exceptionally effective at correcting active metabolic dysfunction 3133. By aggressively lowering circulating glucose, mitigating ectopic lipid deposition, reducing hyperinsulinemia, and suppressing systemic inflammation, metformin removes the accelerated aging drivers inherent to a diabetic state 33326. However, in completely healthy individuals who already possess optimal metabolic flexibility, robust insulin sensitivity, and functional AMPK-mTOR axes, metformin provides rapidly diminishing returns. In fact, by artificially depressing mitochondrial efficiency in a healthy organism, it may inadvertently induce a localized energy crisis that is maladaptive outside the strict context of overnutrition or obesity 312736.
Systematic Reviews in Healthy Demographics
To isolate the drug's effects from disease states, researchers have conducted systematic reviews specifically targeting healthy, non-diabetic populations across diverse demographics. In studies evaluating young adults, non-diabetic elderly outpatients, and obese but metabolically stable women, metformin has demonstrated highly variable effects 3637. While some biomarkers improve - such as a slight reduction in glycated hemoglobin, minor shifts in immune function (e.g., changes in circulating T follicular helper cells), and slight improvements in gait speed among pre-frail older adults - the broad systemic extension of lifespan remains entirely unproven 3137. Body composition changes are equally mixed; while obese women experienced slight reductions in body mass index, healthy young men observed a slight increase in body fat percentage when subjected to metformin regimens 37. Ultimately, the data implies that metformin works primarily by correcting existing dysfunction rather than fundamentally rewriting the aging program in a healthy human body 3136.
The TAME Trial and the Regulatory Evolution of Geroscience
The definitive resolution to the healthy-versus-diabetic longevity debate was architected to be the Targeting Aging with Metformin (TAME) trial 8383940. Designed to observe over 3,000 non-diabetic adults aged 65 - 79 across 14 U.S. clinical research centers for a duration of six years, TAME represents a potential watershed moment in international regulatory science 8313828.
In a historic 2015 decision, the FDA agreed to the trial's unprecedented design, which eschews a traditional single-disease endpoint in favor of a massive composite primary endpoint. This endpoint tracks the onset of multiple age-related comorbidities simultaneously - specifically cardiovascular disease, cancer, cognitive decline (dementia), and all-cause mortality 83139. By formally approving this composite endpoint, the FDA implicitly acknowledged that the biological process of aging itself could be treated as a modifiable clinical indication, effectively creating a regulatory pathway for future longevity interventions to be approved and prescribed strictly for aging 313828.
However, despite this monumental regulatory victory and its profound implications for geroscience, as of April 2026, the TAME trial remains entirely unfunded and has yet to enroll a single patient 383940. The trial has languished in funding purgatory for over a decade. Because metformin is a widely available, off-patent generic drug costing only pennies per dose, there is virtually zero commercial incentive for the pharmaceutical industry to finance a massive, multi-center, multi-million-dollar outcome trial 3139.
The funding deadlock highlights a stark dichotomy in the longevity field: while billions of dollars in private equity flow rapidly into novel, patentable longevity therapeutics (such as cellular reprogramming, advanced GLP-1 agonists, and APOE gene therapies like those seen in Alzheimer's trials), foundational public health initiatives like TAME remain wholly dependent on philanthropic efforts and notoriously slow federal grants 383940. Consequently, the empirical, placebo-controlled proof required to ethically recommend metformin for pure lifespan extension in disease-free humans remains frustratingly absent, leaving clinicians to extrapolate from imperfect epidemiological data 33136.
The Goldilocks Effect: Maladaptive Consequences of Chronic AMPK Activation
While transient AMPK activation is generally heralded as a cornerstone of metabolic health, the kinase operates within exceedingly strict physiological parameters. The biological "Goldilocks effect" dictates that the magnitude, duration, and specific tissue localization of AMPK activation must be perfectly calibrated. Chronic, systemic overactivation of AMPK - whether achieved through aggressive pharmacological intervention or genetic aberration - can unleash severe maladaptive consequences that outweigh any theoretical longevity benefits.
Cardiovascular Risks and Pathological Cardiac Hypertrophy
Historically, pharmaceutical efforts to develop potent, direct, pan-AMPK activators - drugs designed to bind the allosteric ADaM site and activate all possible combinations of the $\alpha$, $\beta$, and $\gamma$ subunits simultaneously across the entire body - have frequently stalled in preclinical development due to severe cardiac toxicity 172943. The human heart expresses a highly specific stoichiometry of AMPK isoforms, predominantly relying on the $\alpha$2, $\beta$2, and $\gamma$2 subunits 117.
While acute, transient AMPK activation in the heart during periods of ischemic stress is profoundly cardioprotective, chronic pharmacological hyperactivation of the specific cardiac $\gamma$2 isoform triggers a devastating cascade. Sustained activation leads to pathological cardiac hypertrophy, a dangerous physical enlargement of the heart muscle that ultimately impairs left ventricular stroke volume and precipitates congestive heart failure 1729. This adverse physical effect is essentially driven by an over-accumulation of cardiac glycogen, a direct consequence of the ceaseless metabolic signal tricking the heart into believing it is perpetually starved for fuel 130. The discovery of this specific $\gamma$2-mediated toxicity forced the rapid abandonment of several early-generation pan-AMPK activators, sharply highlighting the danger of indiscriminately overriding the body's natural energy-sensing checkpoints across all organ systems simultaneously 1743.
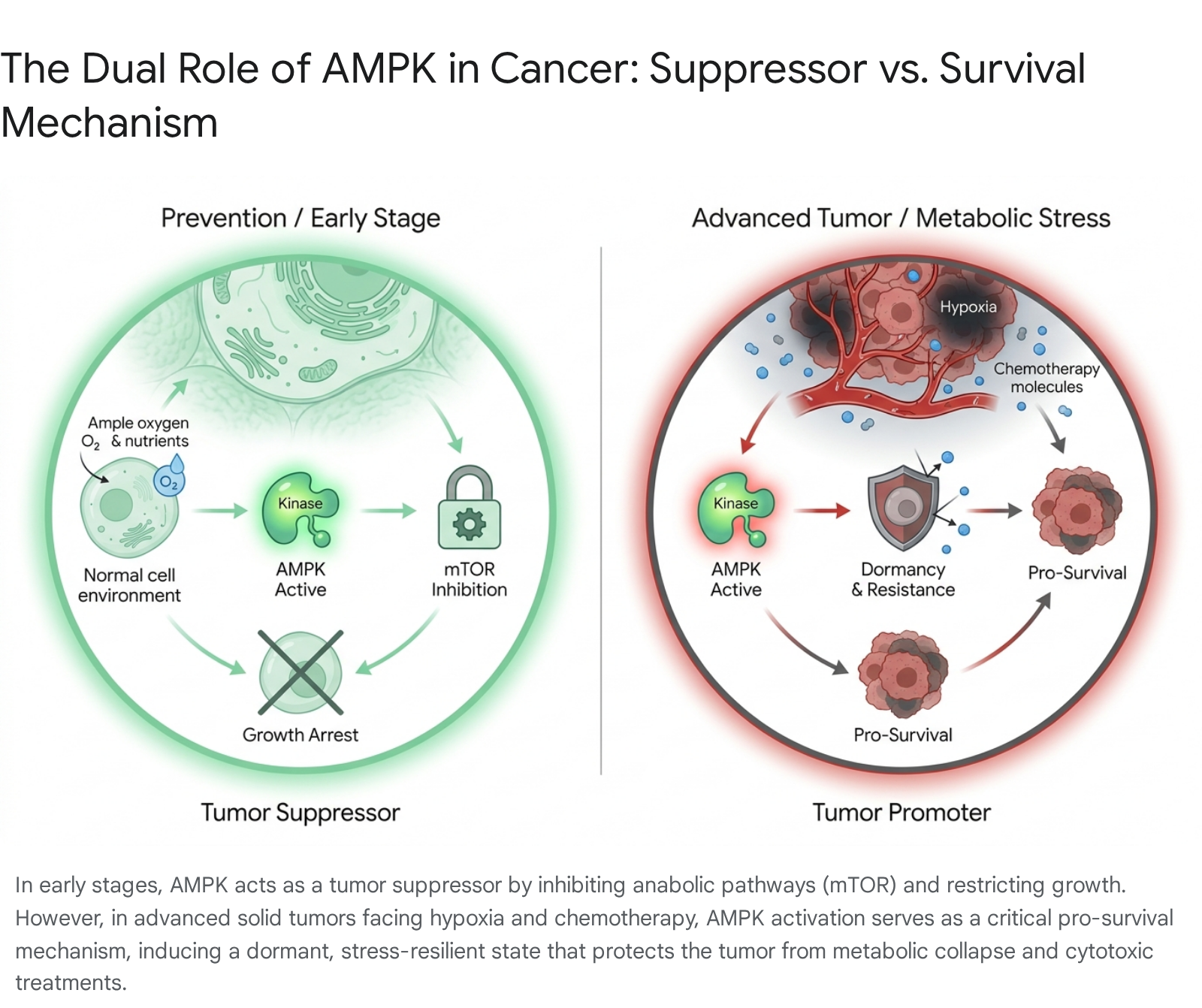
The Contextual Oncogene: AMPK in Cancer Survival
The dual, highly contextual nature of AMPK is most glaringly evident in the field of clinical oncology, where researchers have classified the kinase as a "contextual oncogene" 21731. Initially, AMPK was classified strictly as a potent tumor suppressor due to its close association with the upstream tumor suppressor LKB1. By inhibiting mTORC1, suppressing de novo lipogenesis, and enforcing strict metabolic checkpoints, AMPK restricts the rampant, unchecked anabolic growth characteristic of early-stage carcinogenesis 3531.
However, tumor biology is inherently dynamic. As a solid tumor expands, it inevitably outgrows its localized blood supply, subjecting the inner core of the malignant mass to severe hypoxia, nutrient deprivation, and immense oxidative stress 231. Under these incredibly hostile microenvironmental conditions, AMPK ceases to be a suppressor and switches to act as a potent pro-survival mechanism for the cancer cell 217. By triggering a reversible, non-genetic switch that forces the cancer cell into a dormant, metabolically conservative state - pausing active proliferation while heavily upregulating autophagy to recycle damaged organelles for fuel - AMPK allows the tumor to survive periods of intense stress, including the administration of cytotoxic chemotherapy and ionizing radiation 23132.
Recent 2024 and 2025 human clinical data robustly confirm this pro-tumorigenic role in advanced malignancies. In experiments utilizing patient-derived esophageal adenocarcinoma (EAC) organoids, standard-of-care treatments like cisplatin and ionizing radiation actively triggered rapid AMPK activation 32. Blocking AMPK signaling using specific inhibitors (such as Compound C) or genetic knockdown techniques during chemotherapy drastically sensitized the cancer cells to death, breaking their stress resilience in a highly synergistic manner 232.
Thus, the chronic administration of a potent AMPK activator for anti-aging or longevity purposes carries a massive theoretical risk: if a dormant micrometastasis or undiagnosed early-stage malignancy already exists within an older patient, sustained systemic AMPK activation could inadvertently shield the malignant cells from immune clearance or metabolic starvation, thereby actively promoting tumor survival, progression, and severe drug resistance 21731.
Tissue-Specific AMPK Responses Across Diverse Demographics
The broad application of metabolic modulators is further complicated by diverse demographic variables, including age, biological sex, and baseline metabolic health, all of which fundamentally alter tissue-specific AMPK responses.
Demographic studies consistently show that the natural capacity to activate AMPK in response to exercise or fasting severely blunts with age, contributing heavily to geriatric sarcopenia (age-related muscle loss) and frailty 334849. As DNA damage accumulates with age, the upregulation of DNA repair enzymes like DNA-PK actively inhibits AMPK, creating a vicious cycle of mitochondrial decline and reduced macroautophagy 49.
Biological sex also dictates AMPK responsivity and pharmacological outcomes. In preclinical pain models utilizing AMPK activators, female cohorts exhibited significantly lower pain thresholds than males prior to treatment, yet demonstrated robust gender-adjusted responses to drugs like metformin and ATX-304, highlighting differential neural and inflammatory pathway engagements 34. Furthermore, the presentation of ectopic lipid accumulation differs sharply across demographics; post-menopausal women and older men are significantly more susceptible to visceral and hepatic fat accumulation, increasing their requirement for hepatic-specific AMPK activation to correct dyslipidemia, whereas younger demographics generally require higher muscular AMPK activation to maintain insulin sensitivity 1826. These vast demographic discrepancies underscore why blunt, systemic AMPK activation with older drugs yields highly variable and often contradictory clinical outcomes.
Next-Generation Pharmacotherapy: The Shift to Isoform Selectivity
Recognizing the severe limitations of indirect activators like metformin and the fatal cardiac risks of early pan-activators, the biopharmaceutical industry has aggressively pivoted toward highly selective, direct AMPK activators. By targeting the precise stoichiometric combinations of the $\alpha$, $\beta$, and $\gamma$ subunits expressed in distinct tissues, researchers can successfully isolate the localized metabolic benefits of AMPK activation while entirely circumventing systemic toxicity 172943.
The Direct Pan-Activators: ATX-304 and SCY-770
Direct AMPK activators bypass the mitochondrial electron transport chain entirely. Instead of inducing a localized energy deficit (altering the AMP/ATP ratio) like metformin does, these advanced drugs bind directly to the enzyme complex - typically at the Allosteric Drug and Metabolite (ADaM) site located precisely between the $\alpha$ and $\beta$ subunits 1.
ATX-304 (formerly O304), currently advancing into Phase 2 trials in 2026 under the Cambrian Bio pipeline company Amplifier Therapeutics, is a peripherally restricted pan-AMPK activator 3033345152. Because its molecular structure prevents it from crossing the blood-brain barrier, it entirely avoids overstimulating neurogenesis or triggering central nervous system side effects 5354. In preclinical and early human trials, ATX-304 has demonstrated profound efficacy in shifting the liver's metabolic program to combat Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD). It directly reduces hepatic de novo lipogenesis and increases fatty acid $\beta$-oxidation 34353637. Uniquely, ATX-304 acts as a dual AMPK and mitochondrial activator; it prevents the dephosphorylation of the vital Thr172 residue while simultaneously functioning as a mild mitochondrial uncoupler, thereby successfully avoiding the cardiac glycogen accumulation that doomed previous generations of pan-activators 303536.
SCY-770 (formerly PXL770), originally developed for non-alcoholic fatty liver disease by Poxel SA, represents another major clinical evolution in the space. After missing its primary clinical endpoint in a 12-week Phase 2a trial for liver fat reduction, the highly selective, direct AMPK activator was acquired by SCYNEXIS in early 2026 for up to $196 million 49583839. SCY-770 is now being strategically repurposed to treat Autosomal Dominant Polycystic Kidney Disease (ADPKD), the leading genetic cause of end-stage renal failure 58394041. In ADPKD models, targeted AMPK activation directly suppresses the aberrant proliferation of renal cyst epithelial cells, reduces macrophage infiltration, and heavily downregulates the mTOR-driven cystic growth program, lowering kidney weight ratios by 35% 39. SCYNEXIS anticipates launching a massive Phase 2 proof-of-concept study for SCY-770 in ADPKD patients in the fourth quarter of 2026, with early efficacy readouts expected in the second half of 2027 584063.
The Evolution of True Isoform Selectivity: BLX-0871
Perhaps the most significant and highly anticipated breakthrough in modern AMPK pharmacology is the successful development of true isoform-selective activators, perfectly exemplified by BLX-0871 from Biolexis Therapeutics 1729. The fundamental, unignorable limitation of the current generation of blockbuster weight-loss drugs (such as GLP-1 and GIP receptor agonists) is indiscriminate catabolism: up to 40% of the massive weight lost by patients consists of vital lean skeletal muscle mass. This rapid muscle deterioration permanently damages the patient's resting metabolic rate, promotes frailty, and virtually guarantees rapid weight regain upon cessation of the drug 172942.
Skeletal muscle tissue expresses a highly unique set of AMPK heterotrimers, specifically those containing the $\gamma$3 subunit (the $\alpha$2$\beta$1$\gamma$3 and $\alpha$2$\beta$2$\gamma$3 complexes) 11729. BLX-0871 is an oral, first-in-class small molecule structurally engineered to bind exclusively to these specific $\gamma$3-containing complexes 172943. By strictly ignoring the $\gamma$2 and $\gamma$1 complexes found predominantly in the heart and liver, BLX-0871 triggers "exercise-mimetic" pathways strictly within skeletal muscle and adipose tissue, driving localized glucose uptake and fatty acid oxidation 1743.
In highly anticipated 2025 preclinical data presented at ObesityWeek, BLX-0871, when combined with an oral GLP-1 receptor agonist (BLX-7006), demonstrated exceptional muscle preservation during extreme caloric deficit. The combination allowed for an unprecedented 87% lean mass retention compared to the mere ~50% retention typically seen with standard GLP-1 monotherapies 1729. Furthermore, rigorous GLP toxicology studies confirmed zero cardiotoxicity, proving the efficacy of the isoform-selective design 1743. As BLX-0871 enters Phase 1 human clinical trials in early 2026, it represents the purest application of targeted AMPK pharmacology to date - harvesting the endurance and metabolic benefits of localized AMPK activation while surgically avoiding the cardiac and systemic risks dictated by the Goldilocks effect 172942.
Structured Data Analysis: Comparative Interventions and Tissue Dynamics
To synthesize the complex physiological outcomes, clinical trial data, and advanced pharmacological targeting strategies discussed throughout this report, the following structured tables precisely delineate the interaction dynamics of exercise and AMPK activators, alongside the tissue-specific distribution of AMPK subunits.
Table 1: Comparative Efficacy of Exercise, Metformin, and Next-Gen Activators
| Intervention Modality | Primary Mechanism of Action | Impact on Glycemic Control (HbA1c / FPG) | Impact on Cardiovascular Fitness / Adaptation | Muscle Mass Preservation / Hypertrophy |
|---|---|---|---|---|
| Exercise Alone | High ATP demand increases AMP/ATP ratio; triggers transient mitohormetic ROS. | High efficacy in Prediabetes; Moderate in advanced T2DM. | Optimal. Significant increases in VO2peak, vascular sensitivity, and angiogenesis (via BCL6B). | High. Promotes physiological hypertrophy and prevents geriatric sarcopenia. |
| Metformin Alone | Mild Complex I inhibition; limits ATP production; reduces mitochondrial ROS. | Optimal in advanced T2DM; Moderate in Prediabetes. | Neutral. Lowers resting blood pressure but does not inherently increase VO2 capacity. | Neutral to Slight Catabolic. Inhibits mTORC1; may slightly reduce baseline muscle protein synthesis. |
| Exercise + Metformin | Overlapping AMPK activation; Metformin prematurely quashes adaptive ROS signaling. | Synergistic improvement only in overt T2DM; Inferior to exercise alone in Prediabetes. | Blunted. Significant attenuation of exercise-induced VO2peak, systolic BP improvements, and capillary density. | Blunted. Metformin suppression of mTORC1 and BCL6B limits PRT-induced hypertrophy. |
| BLX-0871 (Phase 1) | Direct, isoform-selective ($\gamma$3) activation via the muscular ADaM site. | High potential. Drives peripheral glucose uptake independent of insulin. | Theoretical exercise-mimetic benefits; successfully avoids cardiac hypertrophy risk. | Exceptional. 87% lean mass retention during massive caloric deficit in preclinical models. |
| SCY-770 (Phase 2) | Direct pan-activation; downregulates mTOR-driven cellular proliferation. | High efficacy in liver (MASLD models); Repurposed for kidney targeting. | Reduces cyst growth and macrophage infiltration in renal tissues. | Neutral. Primary targets are non-muscular (hepatic and renal epithelia). |
Table 2: Tissue-Specific AMPK Isoform Distribution and Pharmacological Targeting
| Target Tissue | Dominant Isoform Expression | Physiological Role of Local AMPK Activation | Clinical Target / Indication | Primary Pharmacological Risk (Overactivation) |
|---|---|---|---|---|
| Skeletal Muscle | $\alpha$2, $\beta$2, $\gamma$3 | Stimulates GLUT4 translocation, fatty acid $\beta$-oxidation, mitochondrial biogenesis. | Obesity, Sarcopenia, T2DM (Targeted by BLX-0871) | Blunting of mechanical hypertrophy if mTORC1 is chronically suppressed post-exercise. |
| Liver | $\alpha$1, $\beta$1, $\gamma$1 | Inhibits de novo lipogenesis and cholesterol synthesis; promotes lipid oxidation. | MASLD, MASH (Targeted by ATX-304) | Potential suppression of necessary hepatic acute-phase responses. |
| Heart | $\alpha$2, $\beta$2, $\gamma$2 | Acute cardioprotection during ischemia; regulates cardiac energy supply. | Historically targeted by early pan-activators (Failed) | Cardiac Hypertrophy, excess glycogen storage, heart failure. |
| Kidney (Epithelium) | $\alpha$1, $\beta$1 | Suppresses mTOR-driven cellular proliferation and fluid secretion in cysts. | ADPKD (Targeted by SCY-770 / PXL770) | Impaired renal filtration dynamics if universally activated. |
| Solid Tumors | Highly Variable | Reversible dormant switch; promotes stress resilience, survival, and autophagy. | Target for Inhibition (Compound C, experimental oncology) | Chemoresistance, survival during severe hypoxia/nutrient deprivation. |
Conclusion
The pursuit of longevity through direct metabolic intervention is rapidly entering an era defined by extreme precision rather than sheer pharmaceutical force. The 2024 - 2026 clinical landscape has unequivocally demonstrated that while metformin remains a foundational, highly effective therapy for reversing the accelerated aging pathologies of type 2 diabetes, its uncritical application in healthy, disease-free individuals - and particularly its simultaneous combination with structured exercise regimens - is fraught with immense physiological compromises 3131618. By chemically suppressing the vital mitohormetic signals generated by reactive oxygen species and deeply repressing transcriptional networks like the BCL6B angiogenic program, metformin actively antagonizes the critical cardiovascular and muscular adaptations that exercise inherently seeks to build 10122421. Furthermore, the ongoing stagnation and funding deadlock of the TAME trial highlights a massive institutional failure to bridge the gap between compelling epidemiological hypotheses and the rigorous, placebo-controlled clinical proofs required for widespread longevity prescription 313940.
Looking forward, the blunt instrument of indirect AMPK activation is being definitively replaced. The discovery of the physiological Goldilocks effect - whereby chronic, systemic AMPK activation risks severe cardiac toxicity and advanced tumor resilience - has paved the way for advanced isoform-selective engineering 21731. Cutting-edge compounds like BLX-0871 and SCY-770, which isolate specific subunits to drive localized tissue benefits while entirely ignoring the heart and other vulnerable organs, represent the pinnacle of current metabolic pharmacology 294340. Ultimately, maximizing human healthspan will not rely on a single, universal metabolic brake, but rather on the intelligent, tissue-specific coordination of cellular energy states tailored to distinct demographic profiles.