Common Misconceptions in Aging Science Research
The translation of aging biology from model organisms to human clinical applications has accelerated over the past decade, formalizing the interdisciplinary field of geroscience. The foundational hypothesis of geroscience posits that targeting the fundamental biological hallmarks of aging - including genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, and recently added dimensions such as impaired macroautophagy, chronic inflammation, and dysbiosis - will simultaneously delay the onset of multiple age-related chronic diseases 12. This paradigm suggests that modulating upstream aging processes is a vastly more efficient medical strategy than the traditional approach of treating individual downstream pathologies like cardiovascular disease or dementia.
While preclinical success in altering the aging trajectories of Caenorhabditis elegans, Drosophila melanogaster, and Mus musculus has been robust, the extrapolation of these findings to human populations has encountered significant empirical friction. A critical appraisal of the longevity field reveals a landscape where enthusiasm frequently outpaces empirical validation. Major scientific, financial, and narrative investments have coalesced around specific interventions and measurement modalities - such as epigenetic aging clocks, off-label pharmaceutical gerotherapeutics like metformin, and senolytic compounds. These investments are frequently premised on assumptions that are currently being challenged by late-stage clinical data, large-scale epidemiological re-evaluations, and deep methodological critiques.
Furthermore, the foundational data informing human longevity, including centenarian demographics and genomic cohorts, exhibit structural biases that compromise their generalizability. This report provides an exhaustive analysis of the largest misconceptions and empirical gaps currently characterizing aging science research, highlighting the necessity for recalibration in how biological age is measured, how therapeutic efficacy is evaluated, and how research capital is deployed.
The Epigenetic Clock Efficacy Gap
The development of epigenetic clocks, algorithms that estimate biological age based on DNA methylation (DNAm) patterns at specific cytosine-phosphate-guanine (CpG) dinucleotides, represents a major technical milestone in molecular gerontology. DNA methylation is a key mechanism of epigenetic regulation that undergoes predictable, spontaneous shifts with age, resulting in site-specific hypermethylation or hypomethylation 34. However, a pervasive misconception in both the scientific community and the commercial longevity sector is that these clocks are universally accurate, interchangeable, and clinically validated surrogate endpoints for mortality and therapeutic efficacy.
Generational Differences in Predictive Power
The assumption that all epigenetic clocks measure the same underlying construct of "biological age" is empirically false. There is a fundamental divergence in the predictive power, mathematical formulation, and biological relevance of different clock generations 566.
First-generation epigenetic clocks, such as the Horvath pan-tissue clock and the Hannum blood clock, were trained via single-step machine learning regression models to predict chronological age based on DNAm profiles 366. The Horvath clock utilizes 353 CpG sites, while the Hannum clock utilizes 71 7. While they exhibit remarkable accuracy in chronological age estimation - frequently achieving high biweight midcorrelation coefficients (0.65 to 0.89) 7 - they perform poorly as granular predictors of mortality or age-related morbidity. Because they were optimized to correlate with the mere passage of time, they filter out the inter-individual biological variance that represents functional aging, phenotypic deterioration, and differential mortality risk 68.
Second-generation clocks, notably PhenoAge and GrimAge, were explicitly trained on clinical biomarkers, healthspan, and mortality outcomes rather than chronological age 36. PhenoAge, developed using data from the NHANES III cohort, utilizes 513 CpG sites correlated with nine clinical biomarkers 3. GrimAge integrates ,1030 CpG sites to optimally predict composite biomarkers of seven DNA methylation proteins and annual smoking pack-years 3. A 2025 large-scale study by Marioni et al., analyzing 18,859 individuals across 174 incident disease outcomes and 10-year all-cause mortality, confirmed that second- and third-generation clocks significantly outperform first-generation clocks in disease settings 9101213. The GrimAge clock has been established as a highly robust predictor of all-cause mortality, earning the moniker of the "death clock" 311.
In statistical analyses performed by researchers at the National Institute on Aging (NIA), GrimAge demonstrated a superior hazard ratio for mortality prediction compared to other epigenetic clocks and traditional telomere length measurements 11.
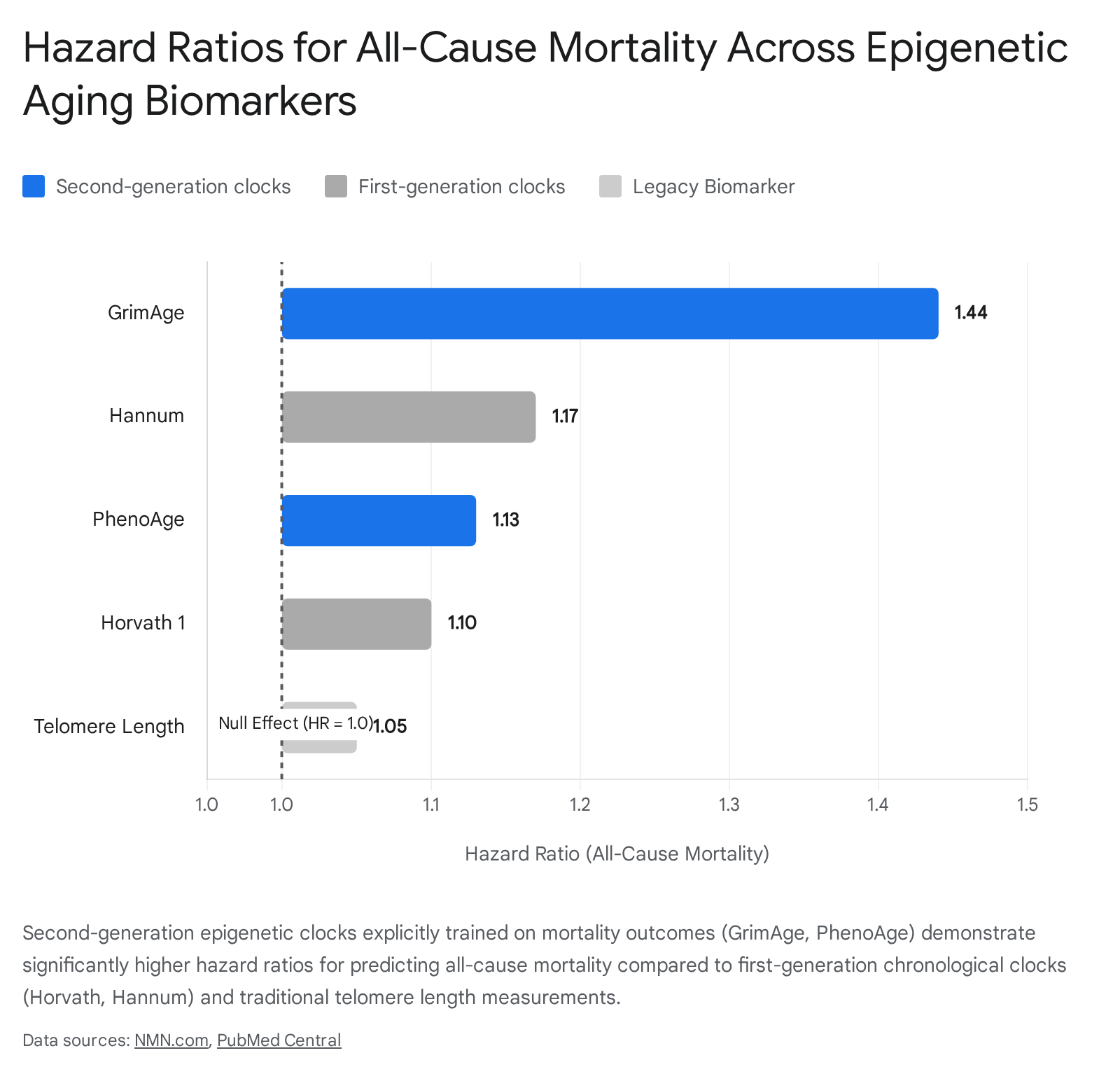
Specifically, each 5-year increase in GrimAge age acceleration is associated with a 44% increased risk of all-cause death, a 33% increased risk of cardiovascular death, and a 54% increased risk of non-cardiovascular death 12.
Despite this evolution, the misconception persists in consumer medicine that first-generation "chronological" clocks can be used to evaluate healthspan interventions. The use of incompatible clocks leads to spurious claims of biological age reversal in commercial environments.
Methodological Limitations and Measurement Errors
Even among advanced epigenetic clocks, significant methodological limitations undermine their utility as definitive individual-level diagnostic tools. The commercial sector frequently markets epigenetic tests as precise, individualized "biological age" calculators, masking the high error rates inherent in the assays 1314.
The median absolute error (MAE) for leading epigenetic clocks remains substantial. For instance, highly optimized deep-learning models like DeepMAge report an MAE of approximately 2.77 years, while traditional elastic net regression models often exhibit MAEs of 3.6 years or higher 31519. This intrinsic computational noise means that an individual testing their biological age twice within a short period could see fluctuations of several years entirely due to stochastic array variance, batch effects, probe hybridization issues, and normalization artifacts rather than any true biological alteration 15.
Furthermore, the generalizability of these clocks across different tissues and age ranges is deeply flawed. A 2025 study evaluating the performance of established adult-trained epigenetic clocks in pediatric cohorts (assessing matched blood and saliva samples from the Kids2Health cohort of 291 children) found that cross-tissue correlations for age acceleration were unacceptably low ($r = 0.20 - 0.68$) 20. The MAEs ranged wildly from 0.95 to over 44 years when adult blood clocks were applied out-of-context, indicating limited generalizability across the life course and across non-homologous tissues 20.
There is also a profound "reference population mismatch" that introduces systemic inequality into biological age readouts. Epigenetic clocks are typically calibrated on populations heavily skewed toward white, socioeconomically advantaged demographics with lower incidences of lifetime adversity 15. When these algorithms are applied to marginalized populations or individuals with high early-life psychosocial adversity, the output often indicates accelerated biological aging 15. It remains highly debated whether this strictly represents modifiable pathological risk or an unalterable epigenetic imprint of structural inequality over which the patient has no control. Regardless, utilizing these scores in clinical settings without adjusting for environmental and demographic contexts risks misinterpreting standard physiological variations as acute mortality threats.
To summarize the functional divergence in epigenetic clock technologies, Table 1 details the distinct characteristics and limitations of the primary models utilized in geroscience.
| Clock Generation | Primary Training Metric | Examples (CpG Count) | Principal Advantages | Key Misconceptions & Limitations |
|---|---|---|---|---|
| First-Generation | Chronological Age | Horvath (353), Hannum (71) | High accuracy in estimating chronological age; operates across varied tissues. | Fails to accurately predict mortality; filters out functional decline; not highly responsive to geroprotective interventions. |
| Second-Generation | Morbidity / Mortality Risk | GrimAge (1,030), PhenoAge (513) | Strongest predictors of all-cause mortality, cardiovascular disease, and healthspan. | Highly dependent on specific biomarkers; susceptible to array variance; high error margins for individual diagnostics. |
| Third-Generation | Pace of Aging (Longitudinal) | DunedinPACE, DunedinPoAm | Measures the rate of aging rather than cumulative age; highly sensitive to lifestyle. | Requires dense longitudinal training data; cross-tissue application is limited; potential overfitting to survivor cohorts. |
| Causal Clocks | Causally linked CpG sites | DamAge, AdaptAge | Attempts to separate DNAm damage from adaptive survival responses. | Does not currently outperform second-generation clocks in hazard ratio prediction; clinical utility remains unproven. |
Reevaluating Metformin as a Universal Gerotherapeutic
Metformin, a biguanide compound utilized globally as a first-line treatment for type II diabetes, has been widely championed over the past decade as the most likely candidate for the first FDA-approved, prophylactic anti-aging drug. Its hypothetical geroprotective mechanisms include the activation of AMP-activated protein kinase (AMPK), the downstream inhibition of the mechanistic target of rapamycin (mTOR) pathway, the suppression of hepatic glucose production, and the mitigation of chronic low-grade inflammation and oxidative stress 1617.
Despite deep mechanistic plausibility and widespread off-label use in longevity circles, the foundational evidence supporting metformin's life-extending efficacy in healthy, non-diabetic individuals is currently facing severe epidemiological and clinical scrutiny.
Methodological Flaws in Observational Epidemiology
The dominant narrative that metformin actively extends human lifespan was heavily catalyzed by highly cited observational studies utilizing the UK Clinical Practice Research Datalink (CPRD). One influential analysis reported that patients with type II diabetes receiving intensive metformin treatment exhibited a 35% reduction in microvascular complications and actually demonstrated better overall survival rates than matched, non-diabetic controls from the general population 161824. This extraordinary claim - that a diabetic patient on metformin could theoretically outlive a healthy individual - sparked a massive paradigm shift in geroscience 24.
However, recent methodological reassessments have identified profound flaws in these early observational cohorts. A 2025 critical review by Keys et al., published in Ageing Research Reviews, dismantles the CPRD survival claim, highlighting severe structural biases - particularly immortal time bias, selection bias, and unmeasured confounding related to diabetes severity 17182419. When subsequent large-scale epidemiological studies attempted to replicate these findings using more robust frameworks, such as the Danish national registers, the survival advantage of metformin users over non-diabetics completely disappeared 1824.
The updated consensus among pharmacoepidemiologists is that while metformin provides significant cardiovascular and mortality benefits relative to other anti-diabetic medications (such as sulfonylureas) within a strict diabetic cohort, it does not elevate a diabetic patient's lifespan above that of a healthy baseline population 2021.
Conflicting Clinical Outcomes and the Necessity of Precision Medicine
Translating the metabolic benefits of metformin to non-diabetic, healthy individuals seeking longevity has yielded equivocal, and sometimes detrimental, results. While some preclinical trials in non-human primates, such as a 2025 Cell study on macaque monkeys, demonstrated a 6-year reduction in brain age and protection against tissue degeneration 22, human trial data remains stubbornly inconsistent 1823.
Clinical trials assessing metformin in older individuals without type II diabetes have highlighted an emerging uncertainty regarding its anti-aging potential. For instance, studies investigating the drug's impact on muscular adaptation during resistance training in older adults found that metformin may actually blunt the hypertrophic response and inhibit exercise-induced improvements in whole-body insulin sensitivity. Because both high-intensity exercise and metformin trigger energetic stress pathways via AMPK, their simultaneous application can result in antagonistic rather than synergistic physiological effects.
Furthermore, the genetic variability of human populations severely complicates metformin's presumed universal efficacy. Preclinical studies using diverse strains of Caenorhabditis elegans demonstrate that while metformin promotes healthy aging in some genetic backgrounds, it has neutral or even negative impacts on lifespan and healthspan in others 24. This highlights a critical misconception in longevity pharmacology: the assumption of uniform drug response. The pursuit of precision medicine dictates that an individual's specific metabolic phenotype, gut microbiome composition, and genetic baseline will dictate whether off-label metformin acts as a geroprotective agent or an unnecessary cellular stressor 1724.
The landmark Targeting Aging with Metformin (TAME) trial, designed to definitively assess whether metformin can delay the onset of a composite of age-related diseases in over 3,000 humans aged 65 to 79, continues to face logistical and funding challenges 162024. While the trial's regulatory design - forcing the FDA to consider aging as a viable primary clinical endpoint - represents a massive policy victory for the field, the scientific expectations for the drug's actual therapeutic efficacy are increasingly tempered by the null findings in recent non-diabetic human literature 1620.
Clinical Translation Hurdles in Senolytic Therapies
Cellular senescence is a fundamental biological state of irreversible cell cycle arrest. Senescent cells alter the tissue microenvironment by secreting a cocktail of pro-inflammatory cytokines, proteases, and chemokines - collectively termed the senescence-associated secretory phenotype (SASP) 3132. The accumulation of these cells over time is a primary driver of tissue dysfunction, fibrotic remodeling, and systemic inflammaging 32. Senolytics, a class of compounds designed to selectively target anti-apoptotic pathways and induce apoptosis in senescent cells, have been widely viewed as the most viable near-term solution for systemic biological rejuvenation 3132.
Preclinical Success Versus Systemic Clinical Realities
The misconception that clearing senescent cells will yield immediate, systemic anti-aging benefits in humans has been harshly challenged by substantial clinical failures. Early preclinical data was immensely promising, demonstrating that interventions like the combination of dasatinib (a repurposed tyrosine kinase inhibitor used in oncology) and quercetin (a plant flavonoid), or fisetin alone, could restore tissue homeostasis, delay frailty, and extend lifespan in both progeroid and naturally aged mice 313225.
However, human clinical translation has proven exceedingly difficult. The most notable setback occurred with Unity Biotechnology, the most heavily capitalized senolytic development company in the sector 31. In August 2020, Unity's lead systemic senolytic candidate, UBX0101 (an MDM2/p53 pathway inhibitor), completely failed to meet primary endpoints in a Phase II clinical trial for osteoarthritis of the knee 3125. The drug was unable to demonstrate statistically significant pain reduction or functional joint improvement compared to a placebo, leading to a massive strategic pivot and workforce reduction 31. Similarly, data from the Interventions Testing Program (ITP) in 2025 contradicted earlier small-scale mouse studies, showing a distinct lack of life extension resulting from fisetin supplementation in robust, genetically diverse mouse models 25.
These failures underscore a fundamental biological reality: the microenvironment of human aging is infinitely more complex than specific, highly controlled murine models of accelerated aging. Senescent cells exhibit profound heterogeneity across different tissues. A senolytic compound that successfully targets the anti-apoptotic Bcl-2 pathway in a senescent adipocyte may have zero efficacy against a senescent endothelial cell or a senescent neuron driving chronic pain 32. The initial expectation of a "broad-spectrum" senolytic pill that systemically clears all aged cells with minimal off-target toxicity is biochemically improbable at this stage of drug development.
The Strategic Shift Toward Disease-Specific Indications
In response to systemic clinical failures, the senolytic field has been forced to pivot away from broad "anti-aging" applications toward localized, disease-specific indications with faster regulatory pathways 31. Unity Biotechnology shifted its pipeline to focus exclusively on ophthalmology, developing UBX1325, a potent senolytic targeting Bcl-xL. This compound is designed for localized injection into the eye to treat diabetic macular edema (DME) and diabetic retinopathy 2526.
Preliminary results in this highly confined, localized environment have been significantly more promising. In the ASPIRE phase 2b study of 52 patients, UBX1325 demonstrated non-inferiority to standard anti-VEGF therapies (aflibercept), achieving comparable visual acuity gains with a durable effect 26. Concurrently, consumer companies like OneSkin have bypassed the FDA pharmaceutical route entirely, successfully launching topical senotherapeutics via cosmetic regulatory pathways, demonstrating targeted reductions of senescent burdens specifically in aged dermal tissue 25.
This pivot highlights a broader reality within geroscience: while the underlying mechanisms of aging are undeniably systemic, the successful pharmaceutical interventions currently in development will likely be tightly localized and disease-specific. The regulatory, financial, and biological hurdles of proving total systemic "rejuvenation" remain far beyond the current capabilities of senolytic pharmacology.
Demographic Homogeneity and Data Anomalies in Longevity Cohorts
A core pillar of aging science is the epidemiological study of populations that exhibit exceptional longevity, specifically centenarians and individuals living in geographically distinct "Blue Zones." The insights derived from these populations shape dietary, genetic, and lifestyle recommendations worldwide. Concurrently, large-scale genomic biobanks are utilized to identify the precise genetic architecture of human lifespan. Both datasets, however, suffer from severe structural biases that the field frequently overlooks.
Statistical Artifacts in Extreme Longevity Data
The validity of demographic data at the extreme tail of the human lifespan is highly contested. A fundamental misconception is that the high density of centenarians in specific global regions is purely the result of optimal genetics, plant-based diets, and low-stress lifestyles. A comprehensive 2024 analysis covering 236 nations and states from 1970 to 2021 revealed that late-life survival data is dominated by systemic anomalies, ascertainment bias, and record-keeping errors 35.
Regions with the highest centenarian attainment rates frequently correlate with poor historical vital registration systems rather than true biological exceptionalism. For example, the top global regions for survival to ages 100+ routinely include nations ranking incredibly low in overall life expectancy - such as Malawi and Thailand (ranking 202nd and 212th, respectively) - as well as territories like Puerto Rico, where historical birth certificates were deemed so unreliable they were legally invalidated as legal documents 35. In many middle-income and developing nations, incomplete death registration and pension fraud further artificially inflate the number of living centenarians 36.
Furthermore, robust clinical characterizations of verified centenarian cohorts challenge the "healthy aging" narrative. The COOLCEN cohort study in Colombia, assessing 160 verified centenarians, revealed that 98% of participants met criteria for frailty or pre-frailty, 75% exhibited sarcopenia, and 63.7% suffered from dementia 36. This data suggests that extreme longevity is frequently characterized by profound morbidity rather than extended healthspan, and implies that many identified "Blue Zones" may be statistical artifacts driven by age exaggeration 3536.
The WEIRD Bias in Genomic Biobanks
When longevity researchers utilize large-scale biobanks to identify genetic markers of aging and train new epigenetic clocks, they rely almost exclusively on WEIRD populations - Western, Educated, Industrialized, Rich, and Democratic 272839. For instance, the UK Biobank, a primary engine for geroscience research and polygenic risk score development, is overwhelmingly homogeneous. Over 85% of its participants identify as White/Caucasian, with disproportionately high levels of tertiary education and socioeconomic status 28.
The consequence of this homogeneity is that genetic risk factors and aging biomarkers derived from these databases do not generalize reliably to the global population 2839. A stark example of this vulnerability is the APOE $\epsilon4$ allele, generally recognized as the strongest genetic risk factor for late-onset Alzheimer's disease and cardiovascular mortality. An analysis comparing outcomes in the UK Biobank to the more diverse All of Us Research Program in the United States revealed highly divergent hazard ratios. While both heterozygous $\epsilon3\epsilon4$ and homozygous $\epsilon4\epsilon4$ genotypes showed significantly higher all-cause mortality risk in the UK Biobank, only the homozygous $\epsilon4\epsilon4$ carriers demonstrated an increased risk in the diverse All of Us cohort 39.
The continued reliance on WEIRD cohorts means that the epigenetic clocks, polygenic risk scores, and mortality predictors currently driving geroscience are inherently tuned to the physiological and socioeconomic baseline of privileged, industrialized populations. Extrapolating these models to a diverse global species without rigorous cross-population validation represents a significant empirical vulnerability 27283929.
Structural Misalignment in Federal Aging Research Funding
The final and perhaps most restrictive misconception regarding the aging field is the public assumption that it receives massive, dedicated institutional funding commensurate with its potential impact on global health. While private sector venture capital has flooded into longevity biotechnology startups (reaching over $5 billion globally), federal public funding is structurally misaligned, heavily favoring downstream disease pathology over upstream basic aging biology 30.
The Dominance of Disease-Specific Funding
The National Institute on Aging (NIA), a branch of the National Institutes of Health (NIH), is the primary federal conduit for aging research in the United States. For Fiscal Year 2025 and 2026, the NIA operates with an appropriated budget in the range of $4.4 billion to $4.5 billion 313233. However, a detailed analysis of the allocation of these funds reveals a deep systemic bias toward end-stage disease rather than preventative geroscience.
More than half of the NIA's awarded grant dollars are strictly dedicated to Alzheimer's disease and Alzheimer's disease-related dementias (AD/ADRD) 34. Through specialized congressional mandates, such as the National Plan to Address Alzheimer's Disease, AD/ADRD receives billions in earmarked funding, including requests for hundreds of millions in additional "Professional Judgment Budgets" annually to expand translational research, clinical interventions, and dementia care models 323536. While this funding is critical for tackling the neurological crises of an aging population, it effectively crowds out research into the root cellular causes of aging that precipitate such neurodegeneration.
The Deficit in Basic Aging Biology Investment
By contrast, the Division of Aging Biology (DAB) - the specific branch of the NIA actually responsible for funding basic geroscience, cellular senescence, mitochondrial dysfunction, epigenetic mechanisms, and the highly successful Interventions Testing Program - receives less than 1% of the total NIH budget 303738. In absolute terms, core aging biology operates on an allocation of roughly $346 million, a mere fraction of the capital deployed toward end-stage neurological decline 3038.
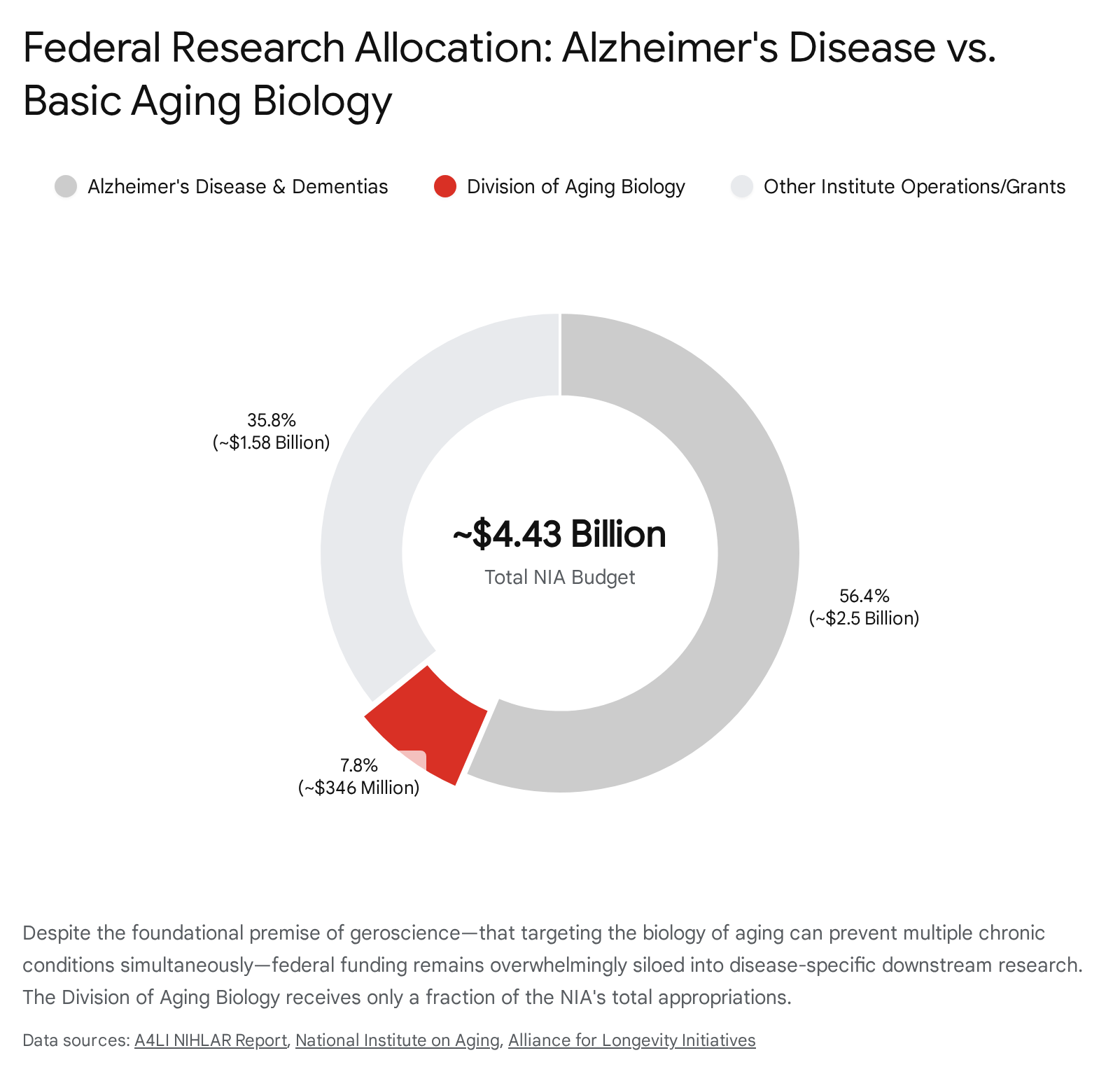
This immense structural discrepancy forces researchers to contort basic longevity and cellular mechanism proposals into Alzheimer's-adjacent frameworks in order to secure grant funding 3940. Advocacy groups within the sector, such as the Alliance for Longevity Initiatives (A4LI), emphasize that this paradigm is reactive rather than preventative 3039. Treating aging as a cohesive biological target requires a fundamental restructuring of federal capital allocation. Proposals currently circulating in policy spheres advocate for consolidating Alzheimer's research into a newly proposed National Institute on Neuroscience, while establishing a separate, dedicated institute (NIHLAR) focused exclusively on aging biology and extending the healthy human lifespan 39. Until this structural funding misalignment is corrected, the public sector's ability to drive systemic, upstream longevity breakthroughs will remain severely constrained by immediate clinical priorities.
Conclusion
The pursuit of human longevity is currently undergoing a necessary maturation phase, transitioning from a period of unbridled optimism to one of rigorous clinical validation. This transition requires the field to identify and discard several pervasive misconceptions.
Biological aging is not yet perfectly quantifiable; current epigenetic clocks exhibit substantial mathematical error rates, reference population mismatches, and generational limitations that preclude their reliable use as definitive individual diagnostics or therapeutic endpoints 61520. Similarly, the off-label use of legacy drugs like metformin and the systemic application of first-generation senolytics are not immediate, universal panaceas. Both therapeutic avenues face high clinical hurdles, complex pleiotropic effects, and a strict requirement for precision-medicine targeting based on individual genetic and metabolic profiles 18243132.
Furthermore, the empirical epidemiological foundations of the field must be cleansed of structural biases, moving away from flawed centenarian record-keeping and homogenous WEIRD genomic cohorts that fail to capture the true diversity of human aging 352839. Ultimately, geroscience holds profound potential to alter the trajectory of human healthspan and alleviate the burden of age-related disease. However, realizing this potential requires an unwavering adherence to empirical reality, an acknowledgment of profound biological heterogeneity, and a structural realignment of global research funding to support the fundamental biology of aging directly.