Effects of GLP-1 receptor agonists on the brain and behavior
Introduction
The clinical implementation of glucagon-like peptide-1 receptor agonists (GLP-1RAs) represents one of the most significant pharmacological developments of the twenty-first century. Initially conceptualized and approved strictly as metabolic interventions to improve glycemic control in patients with type 2 diabetes, these agents mimic the endogenous incretin effect, stimulating glucose-dependent insulin secretion while simultaneously suppressing glucagon release from the pancreas 122. However, their profound efficacy in inducing weight loss has prompted a fundamental reevaluation of their primary mechanism of action. Emerging neuropharmacological data increasingly suggests that the most clinically visible effects of GLP-1RAs - such as dramatic appetite suppression, the reduction of hedonic cravings, and the modulation of generalized reward-seeking behaviors - are driven almost entirely by their interaction with the central nervous system (CNS) 34.
The classification of GLP-1RAs as purely "metabolic" agents is increasingly viewed as a historical artifact stemming from their initial discovery 34. Endogenous GLP-1 is secreted peripherally by enteroendocrine L-cells in the distal gastrointestinal tract in response to nutrient ingestion, but this peripheral hormone possesses a circulating half-life of approximately two minutes due to rapid enzymatic degradation by dipeptidyl peptidase-4 (DPP-4) and subsequent renal clearance 25. Concurrently, GLP-1 is synthesized centrally by preproglucagon (PPG) neurons located in the nucleus tractus solitarius (NTS) of the brainstem, where it functions not as a hormone, but as a spatially precise neurotransmitter 46. The divergence between peripheral hormonal activity and central neurotransmission indicates that modern, long-acting exogenous GLP-1RAs interface with complex neurobehavioral circuits, modulating homeostatic energy balance, dopaminergic reward signaling, and neuroinflammation 67.
Pathways of Central Nervous System Access
A central debate in contemporary incretin pharmacology is precisely how peripherally administered peptides, which are typically large macromolecules, exert such dominant central effects. To achieve therapeutic viability and overcome the two-minute half-life of native GLP-1, pharmacological GLP-1RAs are engineered for extended durability, utilizing structural modifications that incidentally alter their ability to cross the blood-brain barrier (BBB) 58. The current consensus points to two distinct, concurrent pathways through which these agents exert central effects: direct BBB penetration and indirect vagal nerve signaling 7.
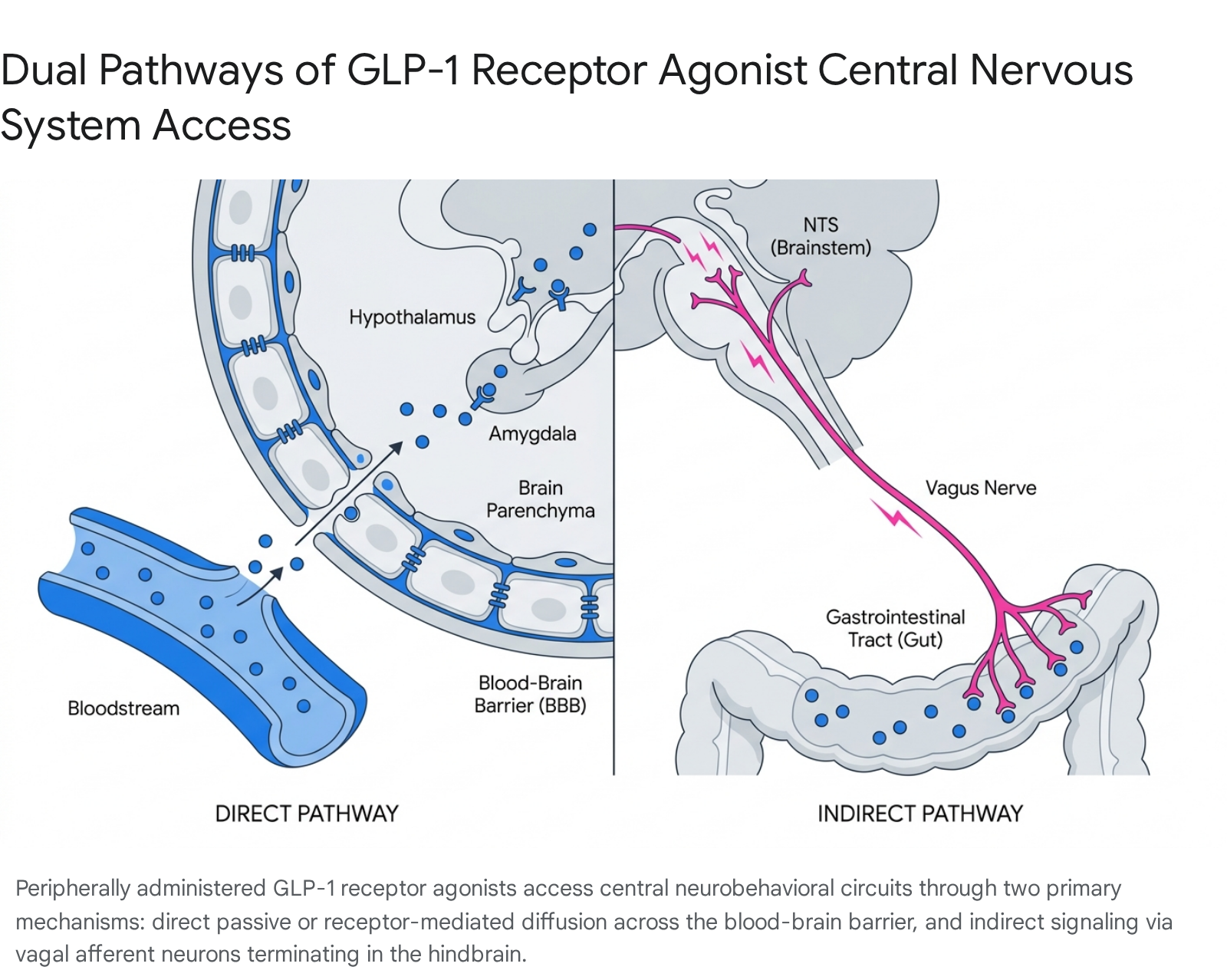
Blood-Brain Barrier Penetration Kinetics
The ability of a GLP-1RA to physically enter brain parenchyma depends heavily on its molecular size, lipid solubility, and engineered modifications 79. Currently available GLP-1RAs are generally divided into two structural classes: exendin-4 derivatives and native human GLP-1 analogs 10.
The first class relies on exendin-4, a native peptide isolated in 1990 from the venom of the Gila monster lizard, which shares structural similarity with human GLP-1 but naturally resists DPP-4 degradation 2. Exendin-4 based agents, such as exenatide and lixisenatide, are non-acylated and demonstrate relatively unhindered passage across the BBB 810. Pharmacokinetic studies utilizing radio-labeled iodine (125I) indicate that exenatide and lixisenatide cross the BBB rapidly via adsorptive transcytosis, a non-saturable process across brain endothelial cells, reaching peak concentrations in the brain parenchyma quickly after administration 81011.
Conversely, the second class consists of long-acting human analogs, such as liraglutide, semaglutide, and dulaglutide. These agents achieve their extended half-lives via acylation - the addition of fatty acid chains that facilitate strong, reversible binding to serum albumin, or through fusion to large carrier proteins like the IgG Fc region 58. While albumin binding protects the peptide from rapid degradation and renal clearance, the massive size of the resulting albumin-peptide complex heavily restricts direct BBB penetration 7.
Liraglutide and semaglutide exhibit significantly lower cerebrospinal fluid (CSF) to plasma ratios compared to shorter-acting non-acylated agents 7. Preclinical kinetics reveal that while liraglutide can eventually be detected in the brain at high doses, it does not cross as readily as exendin-4 13. Studies suggest that highly albumin-bound analogs access the brain primarily through circumventricular organs (CVOs) - specialized regions like the area postrema (AP) and median eminence that possess highly fenestrated capillaries and lack a strict BBB 81012. Additionally, there is evidence that semaglutide may interact with specialized tanycytes lining the ventricular walls, utilizing specialized transport mechanisms rather than broad parenchymal diffusion 210.
Vagal Nerve Signaling and the Gut-Brain Axis
Direct brain penetration is not strictly necessary for GLP-1RAs to exert behavioral and satiating effects 1112. The anatomical gap between the peripheral circulation and the deep central nervous system is effectively bridged by the vagus nerve. GLP-1 receptors are densely expressed on vagal sensory afferent neurons located in the nodose ganglia, which innervate the entire proximal and distal gastrointestinal tract 67.
When peripheral GLP-1 or systemically administered exogenous agonists bind to these vagal afferents, the signal is transmitted directly up the neural axis to the nucleus tractus solitarius (NTS) in the brainstem 37. The NTS serves as a critical integration hub for metabolic signaling. Crucially, it houses the aforementioned preproglucagon (PPG) neurons that synthesize endogenous central GLP-1 6. Activation of this vagal-NTS pathway by peripheral drugs triggers the endogenous release of central GLP-1 directly into deep brain structures, functioning as an internal neurotransmitter cascade rather than relying on the physical diffusion of the exogenous drug into the tissue 34.
The functional dominance of this neural relay is supported by surgical ablation studies. Subdiaphragmatic vagotomy in animal models significantly attenuates the short-term satiation effects and brain activation typically induced by peripheral GLP-1RAs 31013. Furthermore, chemogenetic and optogenetic activation of GLP-1R-expressing vagal sensory neurons in mice reduces food intake for prolonged periods and alters gastric pressure independently of circulating GLP-1 levels 6.
Pharmacokinetic Profiles of Approved and Investigational Agents
To contextualize the varied central effects of these drugs, it is necessary to compare their structural modifications, circulatory half-lives, and theorized mechanisms of brain access. Extended half-lives generally correlate with greater overall systemic exposure and consistent metabolic control, but also align with differing rates of gastrointestinal side effects such as nausea and delayed gastric emptying 5.
| Drug Name | Structural Class | Approximate Half-Life | Primary Brain Access Mechanism | Clinical Indication Focus |
|---|---|---|---|---|
| Exenatide | Exendin-4 derivative | ~2.4 hours | High direct BBB penetration (Adsorptive transcytosis) | Type 2 Diabetes |
| Lixisenatide | Exendin-4 derivative | ~3 hours | High direct BBB penetration (Adsorptive transcytosis) | Type 2 Diabetes |
| Liraglutide | Acylated human analog | ~13 hours | Vagal relay & CVO access (Restricted BBB penetration) | T2D, Obesity |
| Semaglutide | Acylated human analog | ~160 hours | Vagal relay & CVO access (Restricted BBB penetration) | T2D, Obesity |
| Dulaglutide | Fc-fusion human analog | ~120 hours | Limited direct penetration (Large molecular weight) | T2D |
| Tirzepatide | Dual GLP-1/GIP acylated | ~116 hours | Extracellular pathways, slow penetrance | T2D, Obesity |
Data synthesized from pharmacokinetic evaluations and blood-brain barrier transport studies 25789.
Modulation of Reward Circuits and Substance Use
The most profound paradigm-shifting evidence that GLP-1RAs are fundamentally neurological agents stems from their emerging efficacy in treating substance use disorders (SUDs). By dampening the neurochemical processes that drive cravings, these drugs operate on the exact neural circuits implicated in clinical addiction 314.
Mesolimbic Dopamine and Hedonic Regulation
Appetite and consumption are broadly categorized into two neurobiological drives: homeostatic feeding (eating strictly for energy balance, regulated primarily by the hypothalamus and hindbrain) and hedonic feeding (eating for pleasure or reward, regulated by the mesolimbic dopamine system) 1715. GLP-1RAs influence both networks extensively.
Central GLP-1 receptors are highly expressed in the ventral tegmental area (VTA) and the nucleus accumbens (NAc), which serve as the core nodes of the brain's reward and reinforcement circuitry 37. Neurons in the NTS send direct glutamatergic projections to the VTA, establishing an anatomical gut-brain-reward axis 3. When GLP-1 signaling is activated in these mesolimbic regions, it interacts directly with local gamma-aminobutyric acid (GABA) interneurons 319. This GABAergic activation serves to attenuate phasic dopamine release in response to rewarding stimuli 1916.
This dampening effect is not specific to food. The suppression of incentive salience applies universally to highly palatable foods, alcohol, and illicit drugs 316. Functional magnetic resonance imaging (fMRI) in human subjects confirms that GLP-1RAs diminish blood-oxygen-level-dependent (BOLD) responses in the striatum, fusiform gyrus, and anterior cingulate cortex upon exposure to reward-related cues, correlating neurobiologically with a subjective reduction in cravings 216.
Clinical Evidence in Alcohol Use Disorder
The hypothesis that dampening dopaminergic reward signaling can treat addiction has rapidly moved from preclinical animal models to human clinical trials. Large-scale retrospective observational data has repeatedly demonstrated that patients prescribed semaglutide or tirzepatide for metabolic conditions exhibit significantly fewer incident and recurrent diagnoses of alcohol use disorder (AUD) compared to matched cohorts on alternative antidiabetic medications like DPP-4 inhibitors 172218.
These epidemiological signals are now being verified in prospective, randomized controlled trials. In a recent pivotal trial involving 108 treatment-seeking patients with AUD and comorbid obesity, participants were randomized to receive either weekly subcutaneous semaglutide or a placebo, alongside standard cognitive behavioral therapy 1819. Over a 26-week period, participants receiving semaglutide experienced a 41.1% reduction in heavy drinking days, compared to a 13.7% reduction in the placebo group 19.
The number needed to treat (NNT) - a standard clinical metric of efficacy - for semaglutide in this study was calculated at 4.3 19. This substantially outperforms currently approved oral medications for AUD, such as naltrexone or acamprosate, which typically possess an NNT of 7 or higher 1920. While results in individuals with normal body weight remain understudied, the interaction between GLP-1 genetics, baseline adiposity, and alcohol craving is the subject of multiple ongoing Phase 2 trials 1820. Researchers emphasize that there is an urgent need for novel AUD treatments, as no new medications have received regulatory approval for this indication in nearly two decades 20.
Opioid and Stimulant Use Disorder Evidence
The potential utility of GLP-1RAs extends to the opioid epidemic. Historically, pharmacological treatments for opioid use disorder (OUD) have centered on direct opioid receptor modulation (e.g., buprenorphine, methadone) 3. GLP-1RAs offer a novel, orthogonal mechanism by suppressing the downstream dopaminergic reward of the drug rather than blocking the primary receptor 3.
Observational data from a cohort of over 600,000 U.S. veterans with type 2 diabetes revealed that GLP-1 use correlated with a significantly lower risk of developing SUDs involving opioids, cocaine, and cannabis 26. Among those with pre-existing OUD, GLP-1 patients exhibited lower risks of hospitalization, fatal overdose, and suicidal ideation 26. Preclinical evidence is particularly robust for fentanyl and heroin; GLP-1 receptor activation consistently reduces drug self-administration and prevents cue-induced reinstatement of drug-seeking behavior in rodent models 322. Currently, semaglutide is being formally tested in Phase 2 trials as an adjunct to buprenorphine or methadone maintenance to prevent illicit opioid use 21.
Ongoing Clinical Trials in Addiction Medicine
The transition of GLP-1RAs into addiction medicine is moving at a rapid pace, with dozens of trials currently registered globally 2622. The table below summarizes several critical ongoing clinical trials evaluating these agents for various substance use disorders.
| Clinical Trial Identifier | Investigational Agent | Target Substance | Phase | Primary Endpoint Focus | Status / Expected Completion |
|---|---|---|---|---|---|
| NCT05895643 | Semaglutide (2.4 mg) | Alcohol Use Disorder | Phase 2 | Change in heavy drinking days | Expected Late 2025 18 |
| NCT07218354 | Semaglutide (2.4 mg) | Alcohol Use Disorder | Phase 2/3 | Timeline Follow-Back (TLFB) drinking reduction | Expected 2028 20 |
| NCT06548490 | Semaglutide (1.0 mg) | Opioid Use Disorder | Phase 2 | Illicit opioid use reduction on MOUD | Recruiting 21 |
| NCT07420283 | Brenipatide (Dual) | Opioid Use Disorder | Phase 2/3 | Weeks of abstinence via urine drug screen | Expected 2028 2923 |
| NCT04199728 | Liraglutide (3.0 mg) | Opioid Use Disorder | Phase 2 | Reduction in acute opioid craving metrics | Recruiting 24 |
Structural Innovations and Next-Generation Agonists
The evolution of incretin therapies is shifting away from single-target, injectable macromolecules toward small-molecule oral agents and multi-receptor co-agonists. These structural changes fundamentally alter how the drugs interact with human physiology and, specifically, how they penetrate the brain.
Small-Molecule GLP-1 Receptor Agonists
The advent of non-peptidic, small-molecule GLP-1RAs, such as orforglipron and the recently discontinued danuglipron, marks a critical pharmacological shift in metabolic and neurological medicine 152526. Unlike large peptide analogs that bind broadly to the extracellular domain (ECD) of the GLP-1 receptor, small molecules bind exclusively within a non-canonical orthosteric pocket deep within the receptor's transmembrane (TM) domain 2627.
This unique transmembrane binding mechanism results in "biased agonism" 26. Native peptides and large analogs typically activate both the stimulatory G-protein (Gs) signaling pathway and recruit β-arrestin 2628. Small molecules, however, preferentially activate the Gs signaling pathway while exhibiting minimal recruitment of β-arrestin 2628. Because β-arrestin is the primary mediator of receptor internalization, desensitization, and many gastrointestinal adverse effects, bypassing this pathway results in more sustained receptor signaling and a highly modified tolerability profile 2628.
Crucially, due to their low molecular weight and high lipophilicity, small-molecule GLP-1RAs exhibit deep and unhindered penetration of the blood-brain barrier 1029. Recent neuroanatomical mapping studies utilizing gene-edited mice revealed that these oral small molecules directly activate neurons in the central amygdala (CeA) - a deep brain region closely associated with emotional desire and reward integration, which large peptide agonists struggle to reach directly 1519. Activation of GLP-1 receptors in the CeA was shown to directly suppress dopamine release in response to hedonic feeding, establishing a novel, deeply centralized neural circuit for small-molecule efficacy that operates independently of the vagal hindbrain relay 1715.
Dual and Triple Receptor Co-Agonists
To maximize systemic and neurological efficacy, modern pharmacology has successfully combined GLP-1 agonism with other gastrointestinal hormone targets, primarily glucose-dependent insulinotropic polypeptide (GIP) and glucagon (GCG) 930.
Tirzepatide, a dual GLP-1/GIP receptor agonist, currently demonstrates superior weight loss (averaging up to 20-22%) and superior glycemic control compared to selective GLP-1RAs like semaglutide 383132. Tirzepatide's larger structural configuration limits its direct BBB penetrance; studies indicate it does not enter the brain rapidly within the first hour of administration, but rather reaches the brain parenchyma slowly, likely via extracellular pathways 933. However, its synergistic effects on peripheral metabolism and central satiety networks yield profound clinical results.
Brenipatide (LY-3537031) is an investigational dual GLP-1/GIP agonist currently moving rapidly through Phase 2 and 3 trials specifically designed for non-metabolic, neurobehavioral indications. Current major trials for brenipatide include evaluations for opioid use disorder, alcohol use disorder, and major depressive disorder 292334. The targeted development of brenipatide for psychiatric and addiction indications underscores the pharmaceutical industry's recognition that incretin polypharmacology acts extensively on the central nervous system, attempting to create a "pipeline in a product" that spans both immunology and neurology 3435.
Similarly, mazdutide - a dual GLP-1/GCG receptor agonist - is currently in the Phase 3 LIGHT-COG trial (NCT07083154) to evaluate its specific disease-modifying effects on cognitive dysfunction in patients with type 2 diabetes and early dementia 36. Preclinical studies in metabolic disease models show that mazdutide significantly improves cognitive performance and neuronal structure compared to GLP-1 monotherapy 37. By combining GLP-1's neuroprotective signaling with glucagon's stimulatory effects on energy expenditure and synaptic plasticity, dual agonists target multiple axes of central metabolic dysregulation simultaneously 37.
Neuroprotective Applications in Neurodegenerative Disease
Beyond modulating reward, satiety, and addiction circuits, GLP-1R signaling exerts profound neurotrophic and neuroprotective effects. The activation of central GLP-1 receptors upregulates several critical intracellular survival pathways - most notably the cAMP/PKA, PI3K/Akt, and MAPK cascades 7. Collectively, these pathways inhibit neuronal apoptosis, reduce oxidative stress, and shift microglial cells from a pro-inflammatory state toward an anti-inflammatory, neuroprotective phenotype 7. This robust cellular profile has catalyzed multiple large-scale clinical trials evaluating GLP-1RAs as disease-modifying treatments for irreversible neurodegenerative conditions 1247.
Alzheimer's Disease Efficacy and Trial Discrepancies
The potential for GLP-1RAs to alter the trajectory of Alzheimer's disease (AD) is currently heavily debated due to recent, highly conflicting clinical trial outcomes. The rationale for these trials is strong: retrospective cohort data has repeatedly shown that patients with type 2 diabetes taking GLP-1RAs face a significantly reduced risk of developing all-cause dementia and AD compared to those on other antidiabetic drugs 3338.
The phase 2b ELAD trial evaluated the daily subcutaneous administration of liraglutide over 12 months in patients with mild-to-moderate AD who did not have diabetes. While the trial technically missed its primary endpoint regarding improvements in cerebral glucose metabolism (measured via FDG-PET), structural MRI outcomes were striking. Liraglutide reduced hippocampal volume loss by nearly 50% and attenuated cognitive decline by 18% compared to the placebo group 394041. Liraglutide's specific ability to act on vagal afferents and access key circumventricular nodes is hypothesized to reduce neuroinflammation sufficiently to slow cortical atrophy 710.
In stark contrast, the highly anticipated Phase 3 EVOKE and EVOKE+ trials - which tested daily oral semaglutide in over 3,800 patients with early symptomatic AD - were terminated early after failing to show any clinical efficacy 3852. Semaglutide failed entirely to differentiate from placebo on the primary endpoint of the Clinical Dementia Rating - Sum of Boxes (CDR-SB), showing no ability to slow cognitive or functional decline over two years 383952.
The divergence in these results may stem directly from the differing pharmacokinetics and structural properties of the drugs. Liraglutide possesses a much shorter half-life and a different central penetration profile than semaglutide. Furthermore, the robust central effects seen in the ELAD trial utilizing a direct subcutaneous injection may not translate to the distinct systemic exposure profile generated by daily oral semaglutide, which relies on a specialized sodium caprate (SNAC) absorption enhancer that yields roughly 1% oral bioavailability 72938.
Parkinson's Disease and Motor Symptom Preservation
The clinical outlook for GLP-1RAs in Parkinson's disease (PD) is currently considerably more optimistic than in AD. The underlying pathophysiology of PD features severe neuroinflammation, mitochondrial dysfunction, and the progressive apoptosis of dopaminergic neurons in the substantia nigra - all pathological mechanisms that central GLP-1 signaling actively combats in preclinical models 730.
The LIXIPARK Phase 2 trial, published in The New England Journal of Medicine in 2024, evaluated the short-acting agent lixisenatide in 156 patients with early Parkinson's disease across 21 research centers in France 4243. Over a 12-month period, patients receiving daily subcutaneous lixisenatide (in addition to their standard dopaminergic therapy) showed virtually no progression in motor disability, whereas the placebo group experienced steady, continuous clinical deterioration 424445.
The difference in the Movement Disorder Society Unified Parkinson's Disease Rating Scale (MDS-UPDRS) motor scores between the groups was statistically significant. Crucially, this divergence persisted even after a two-month drug washout period, strongly implying true structural disease modification rather than mere symptom masking 424345. However, researchers noted that lixisenatide did trigger higher rates of gastrointestinal side effects compared to placebo, and it did not improve non-motor symptoms or overall quality of life metrics 4445.
The clinical success of lixisenatide in PD highlights a critical pharmacological principle. Lixisenatide is a non-acylated exendin-4 derivative known for high, unhindered BBB penetrance via adsorptive transcytosis 813. This implies that therapeutic efficacy in deep-brain neurodegenerative disease likely requires molecules optimized for direct, high-volume CNS exposure 47. Long-acting, highly albumin-bound agents like semaglutide may be optimal for peripheral metabolic control and vagal-mediated satiation, but short-acting, brain-penetrant agents may ultimately prove superior for direct neuroprotection.
Conclusion
The initial medical categorization of GLP-1RAs as strictly "metabolic" or "antidiabetic" drugs is conceptually incomplete. While their downstream effects result in profound systemic metabolic improvements - such as lowered hemoglobin A1c, drastically reduced adiposity, and improved cardiovascular outcomes - the upstream mechanisms driving these changes are overwhelmingly neurological 311.
The suppression of appetite relies heavily on the complex interplay between the gut-brain axis, the vagus nerve, the nucleus tractus solitarius, and the arcuate nucleus of the hypothalamus 725. Furthermore, the blunting of cravings, the reduction in heavy alcohol consumption, and the suppression of illicit drug-seeking behaviors are mediated by deep midbrain structures, including the ventral tegmental area, the nucleus accumbens, and the central amygdala 719.
Therefore, GLP-1RAs and emerging dual incretin agonists should be conceptualized fundamentally as broad-spectrum neurobehavioral modulators that happen to possess peripheral insulinotropic properties. As structural innovations continue to shift toward small molecules with deep brain penetrance and dual-agonists that target multiple neural pathways, the utility of these agents will likely expand far beyond endocrinology, embedding them permanently into the therapeutic armamentarium of psychiatry, addiction medicine, and neurology.