Neurobiology of hunger and appetite regulation
Appetite control and the regulation of energy homeostasis represent a highly complex psychobiological system, driven by synchronous interactions between the central nervous system, the gastrointestinal tract, and adipose tissue. Far from being a simple matter of volumetric capacity or conscious willpower, the decision of when eating begins and terminates is governed by a sequence of anticipatory, orosensory, gastric, and enteroendocrine signals. Recent advancements in metabolic research, neurobiology, and precision pharmacotherapy have fundamentally shifted the clinical understanding of human feeding behavior, mapping the specific neuronal microcircuits that dictate hunger and fullness. This report provides an exhaustive analysis of the physiological mechanisms regulating satiety, the dynamic function of the gut-brain axis, the biological phenomena of metabolic adaptation, and the disruptive impact of modern ultra-processed food environments.
Temporal Dynamics of Satiation
The feeding behavior of animals is an adaptive response arising from the demands of the internal environment, modulated by limitations imposed by external availability. Feeding behavior is structured around two distinct concepts: satiation, the dynamic intra-meal cascade of processes that lead to the termination of eating, and satiety, the subsequent post-meal physiological state that suppresses hunger and inhibits further consumption for a given period 12. The onset of satiation is not delayed until nutrients are absorbed; rather, it is initiated by sensory anticipation and executed through an organized behavioral sequence.
Pre-Ingestion Anticipation and Cephalic Responses
The processes leading to meal termination begin prior to the first mechanical bite of food. When an individual plans a meal or is exposed to food-related visual and olfactory stimuli, cortical and brainstem regions are immediately activated 13. This anticipation initiates the cephalic phase response, a preparatory state wherein the brain primes the digestive system. The nucleus tractus solitarius (NTS), located in the brainstem, integrates these sensory cues and relays signals to the dorsal motor nucleus of the vagus nerve. This triggers the early secretion of saliva, digestive enzymes, and a preliminary release of insulin even before nutrients enter the systemic circulation 3.
At the neurological level, the anticipation of food rapidly alters the activity of key feeding circuits in the arcuate nucleus of the hypothalamus (ARC). Within seconds of smelling or seeing food, hunger-promoting Agouti-related peptide (AgRP) neurons are uniformly inhibited, while pro-opiomelanocortin (POMC) neurons are activated 34. The olfactory cortex supplies indirect excitatory and inhibitory inputs to these subsets of appetite-linked neurons 4. This anticipatory inhibition of AgRP neurons is proportional to the expected energy content and palatability of the food, suggesting that the brain relies on learned associations between sensory cues and caloric value to preemptively modulate the drive to consume 34. Recent in vivo fiber photometry studies combining continuous arterial glucose monitoring revealed that this anticipatory "first phase" of AgRP neuronal inhibition is a learned response, occurring prior to nutrient delivery when glucose is administered orally, but failing to occur when glucose is injected intraperitoneally 557.
Orosensory Feedback and the Behavioral Satiety Sequence
As ingestion begins, taste buds and mechanoreceptors in the oropharyngeal area act as the first direct physiological monitors of caloric intake 3. Recent neurobiological mapping has identified specific subsets of brainstem cells - prolactin-releasing hormone (PRLH) neurons - that react to the perception of flavor within seconds to curtail food intake 6. Stimulated by the taste of food, PRLH neurons leap to attention, effectively pacing the rate of consumption to prevent the individual from eating so rapidly that the slower gastrointestinal signals are outpaced 6. This immediate orosensory feedback creates an essential feed-forward loop, balancing the hedonic drive of palatable food with a neural braking system that monitors feeding velocity. The activity of these PRLH neurons also influences subjective palatability, providing a biological basis for the human experience of sensory-specific satiety, wherein food becomes less appetizing as one reaches capacity 6.
In tandem with neural signaling, eating initiates a structured behavioral pattern known as the Behavioral Satiety Sequence (BSS). Observed consistently across mammalian species, the BSS outlines the transition from active feeding to post-meal behaviors. Following the consumption of food, an animal reliably transitions into a period of grooming and general locomotor activity, which eventually culminates in resting or sleeping 2. Pharmacological interventions that induce nausea or malaise disrupt this natural sequence, whereas true satiety-inducing agents, such as cholecystokinin octapeptide (CCK-8S), preserve and accelerate the natural progression from eating to resting 2.
Gastrointestinal Signalling and Mechanoreception
Following mastication and swallowing, food enters the gastrointestinal tract, shifting the regulatory burden from rapid sensory neural pathways to a combination of mechanical vagal feedback and slower peptide hormone secretion.
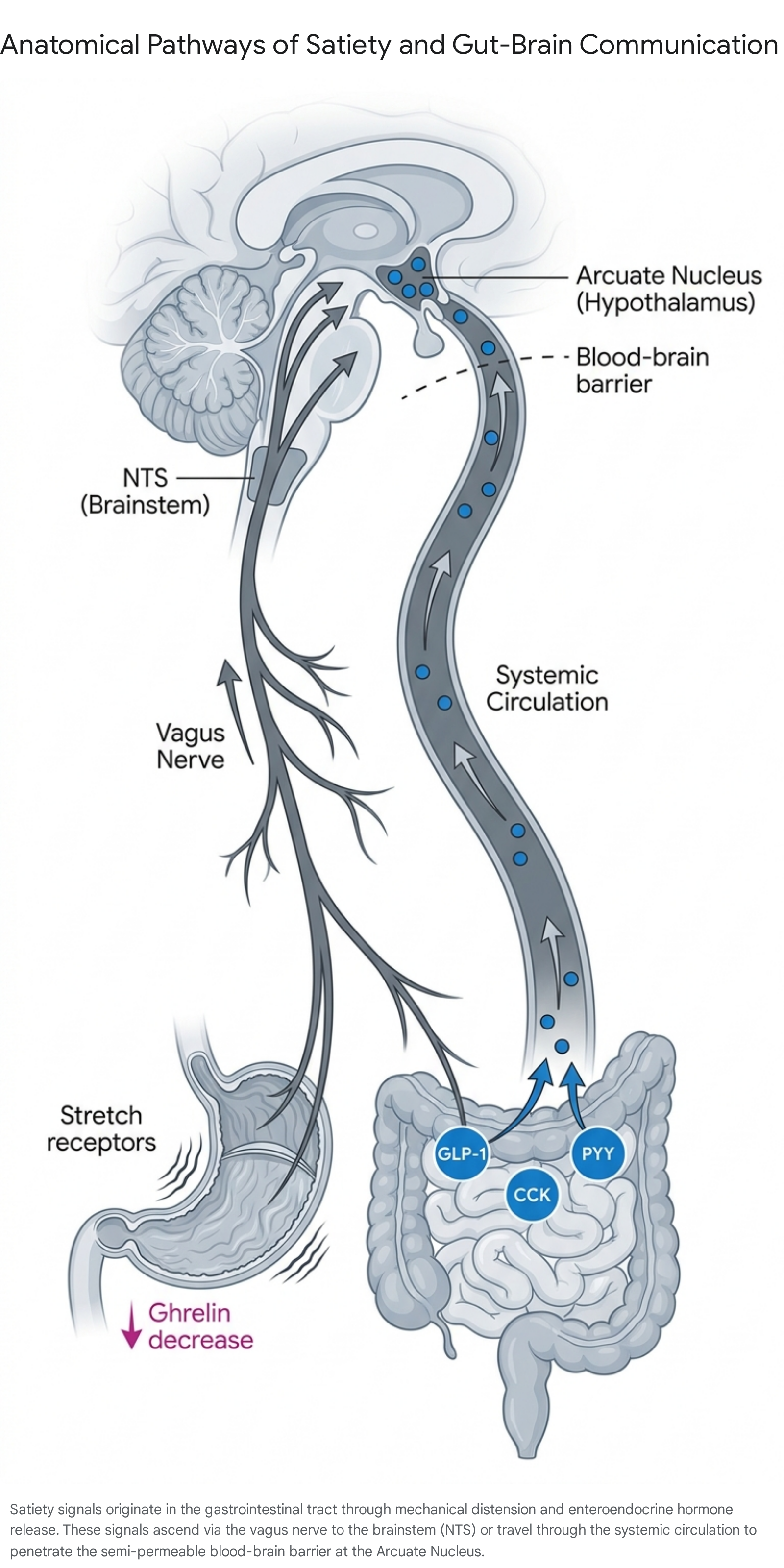
Gastric Accommodation and Distension
The entry of food into the stomach initiates the vagovagal reflex, resulting in a physiological response known as gastric accommodation. This reflex entails a transient relaxation of the proximal stomach, specifically the gastric cardia, fundus, and body, allowing the stomach to receive a volume of food without an immediate, sharp spike in pressure 37. This brief decrease in intragastric pressure at the onset of a meal is a major determinant of overall meal volume 7.
As food accumulates and mixes with gastric juices to form chyme, intragastric pressure gradually rises. Tension-sensitive mechanoreceptors embedded in the gastric walls are stimulated by this distension. These receptors transmit signals via specific vagal afferent nerves (VANs) - such as VANOxtr+ and VANGLP-1R+ fibers - through the nodose ganglion to the NTS in the brainstem 3. This mechanical stretch provides the earliest visceral sensation of physical fullness, prompting the subjective feeling of satiation long before nutrients are fully digested or absorbed into the systemic circulation 38.
Enteroendocrine Nutrient Sensing
As gastric emptying accelerates, chyme is pushed into the duodenum and jejunum, exposing the intestinal lining to a concentrated mix of macronutrients. Enteroendocrine cells distributed throughout the intestinal epithelium act as chemosensors, detecting the specific nutrient profile of the meal. In response, they release a suite of short-term, appetite-suppressing peptide hormones into the local tissue and systemic circulation 311.
Concurrently, the stomach rapidly ceases the production of ghrelin, an orexigenic (appetite-stimulating) hormone synthesized by the gastric mucosa during fasting 311. Ghrelin levels peak in the hour preceding a meal and plummet rapidly upon feeding, providing a sharp negative feedback signal that the immediate energy deficit has been addressed 11. The interaction of these hormones establishes a chemical blockade against continued feeding.
| Hormone | Primary Source | Stimulus for Release | Mechanism of Action |
|---|---|---|---|
| Ghrelin | Gastric mucosa | Fasting, empty stomach | Stimulates AgRP/NPY neurons, reduces energy expenditure 11910. |
| Cholecystokinin (CCK) | I-cells (Duodenum, Jejunum) | Dietary fats and proteins | Slows gastric emptying, signals via CCK-1 receptors on vagal afferents 1110. |
| Glucagon-like Peptide-1 (GLP-1) | L-cells (Distal ileum, colon) | Nutrient transit (carbohydrates/fats) | Enhances glucose-dependent insulin secretion, slows motility, central anorexigenic effect 31110. |
| Peptide YY (PYY) | L-cells (Ileum, colon) | Caloric load in distal intestine | Inhibits NPY/AgRP circuits, suppresses appetite post-meal 111011. |
| Pancreatic Polypeptide | F-cells (Pancreas) | Caloric intake | Slows gastric emptying, inhibits NPY/AgRP system 10. |
| Amylin | $\beta$-cells (Pancreas) | Feeding, rising glucose (co-released with insulin) | Slows gastric emptying, selectively signals POMC neurons 1012. |
Hypothalamic Integration of Energy Status
While the brainstem processes immediate, short-term satiety signals, the arcuate nucleus (ARC) of the hypothalamus acts as the central command center for long-term energy homeostasis. Situated adjacent to the third ventricle, the ARC features a specialized, semi-permeable blood-brain barrier. This structural characteristic allows circulating hormones and macronutrients - such as glucose, amino acids, and free fatty acids - to directly interact with first-order neurons, bypassing traditional central nervous system gatekeeping 310.
The Arcuate Nucleus Microcircuits
The ARC contains two distinct, functionally antagonistic neuronal microcircuits that integrate peripheral signals to dictate feeding behavior: the AgRP/NPY neurons and the POMC neurons.
Neurons co-expressing Agouti-related peptide (AgRP) and Neuropeptide Y (NPY) are the primary drivers of hunger and energy conservation 910. During states of caloric deficit or fasting, these neurons exhibit enhanced action potential firing and spontaneous subthreshold currents 13. Their activation stimulates voracious food-seeking behavior, promotes lipogenesis, and decreases energy expenditure to protect the organism from starvation 34. The orexigenic effect of AgRP depends largely on the co-release of NPY and gamma-aminobutyric acid (GABA), which establish direct monosynaptic inhibitory control over the satiety-promoting POMC neurons. This architecture ensures that when the body requires energy, the satiety pathways are aggressively silenced 31113.
Conversely, Pro-opiomelanocortin (POMC) neurons represent the primary anorexigenic circuit within the ARC. When stimulated, POMC is cleaved to produce alpha-melanocyte-stimulating hormone ($\alpha$-MSH). The release of $\alpha$-MSH activates melanocortin-3 and melanocortin-4 receptors (MC3R/MC4R) in downstream hypothalamic regions, such as the paraventricular nucleus (PVN), leading to profound appetite suppression and an increase in energy expenditure 10. POMC neurons are robustly activated by signals indicating energy surplus, most notably leptin - an adipokine secreted in proportion to total body fat mass. Leptin gradually increases the firing rate of POMC neurons while simultaneously inhibiting AgRP neurons, creating a dual mechanism to limit long-term food intake .
| Feature | AgRP / NPY Neurons | POMC Neurons |
|---|---|---|
| Primary Function | Orexigenic (promotes hunger and food intake) | Anorexigenic (promotes satiety and cessation of eating) |
| Activated By | Ghrelin, energy deficit, fasting, sensory food anticipation | Leptin, GLP-1, PYY, Amylin, Insulin, high energy stores |
| Inhibited By | Leptin, Insulin, GLP-1, GIP, glucose | AgRP-derived GABA release, fasting, Ghrelin |
| Key Neuropeptides | Agouti-related peptide (AgRP), Neuropeptide Y (NPY), GABA | Alpha-melanocyte-stimulating hormone ($\alpha$-MSH) |
| Downstream Target | Antagonizes MC3R/MC4R; inhibits PVN satiety pathways | Agonizes MC3R/MC4R; activates PVN satiety pathways |
Incretin Hormone Dynamics
Recent neuropharmacological investigations, particularly advanced studies published in 2025 and 2026, have significantly refined the understanding of how both endogenous hormones and novel weight-loss drugs manipulate the ARC microcircuits. Analogs of incretin hormones, specifically GLP-1 receptor agonists (e.g., semaglutide) and dual GLP-1/GIP receptor agonists (e.g., tirzepatide), have revolutionized obesity pharmacotherapy.
While both hormones suppress appetite, a 2025 study utilizing in vivo fiber photometry revealed distinct physiological roles. The researchers discovered that endogenous glucose-dependent insulinotropic peptide (GIP) - not GLP-1 - is necessary for the normal, nutrient-mediated inhibition of hunger-promoting AgRP neurons 14. To suppress appetite naturally, GIP receptor activation transmits critical gut-brain communication that silences AgRP neurons. By contrast, at pharmacologic, supratherapeutic doses, both GIP and GLP-1 analogs are sufficient to rapidly inhibit AgRP neuronal dynamics, establishing a "double whammy" effect that suppresses hunger and prevents the rebound hyperphagia normally triggered by weight loss 1415. Furthermore, research indicates that the overconsumption of sucrose (a high-sucrose diet) pathologically alters these interoceptive circuits, specifically blunting the silencing response of AgRP neurons to intragastrically delivered glucose and endogenous incretins 1420.
Amylin and ERK Signaling Pathways
Amylin, a hormone co-secreted with insulin from pancreatic $\beta$-cells in response to feeding, has emerged as another critical satiety regulator. Recent cellular analyses demonstrate that amylin selectively acts on POMC neurons - not NPY neurons - in the arcuate nucleus. Amylin phosphorylates extracellular signal-regulated kinase (p-ERK) almost exclusively within POMC-expressing cells, enhancing the development of $\alpha$-MSH axonal projections to the paraventricular nucleus 1221.
The action of amylin is highly dependent on receptor activity-modifying proteins (RAMP1/3). In mice deficient in RAMP1/3, the density of $\alpha$-MSH fibers decreases significantly, impairing the core satiety network 1221. Furthermore, amylin exerts an additive, synergistic effect with leptin, mediated by STAT3 phosphorylation in the ventromedial nucleus, to heavily suppress food intake 1216. This represents a highly specific, targeted anorexigenic pathway independent of standard incretin signaling, reinforcing the multi-layered defense the brain utilizes to monitor caloric intake.
The Microbiota-Gut-Brain Axis
Beyond direct endocrine and vagal reflexes, the gut microbiome has been recognized as an indispensable third channel in the gut-brain axis, functioning through interconnected neural, immune, and metabolic pathways 1718. The intestinal flora communicates with the central nervous system via the production of microbially derived molecules, most notably short-chain fatty acids (SCFAs) such as butyrate, propionate, and acetate, which are generated through the bacterial fermentation of dietary fibers 1718.
Microbial Metabolites and Enteroendocrine Modulation
SCFAs exert direct influence on enteroendocrine cells. For example, propionate, produced by the microbial fermentation of prebiotics like beta-glucan, specifically stimulates L-cells to increase the secretion of GLP-1 and PYY, effectively amplifying the host's natural satiety signals 1819. Secondary bile acids (2BAs) and tryptophan metabolites produced by the microbiome similarly interact with neuroendocrine receptors to modulate hunger 1718. Dysbiosis - an imbalance in the microbial community structure - can impair SCFA production, leading to localized gut inflammation, reduced GLP-1 secretion, and impaired systemic satiety signaling, which correlates with the onset of metabolic and eating disorders 19.
Neuroimmune Pathways and Vagal Serotonin
The gut microbiome is heavily implicated in neurotransmitter synthesis. Specialized enteroendocrine cells in the gut wall synthesize approximately 95% of the body's serotonin 20. Emerging evidence indicates that microbial metabolites heavily regulate this synthesis, with serotonin subsequently binding to vagal sensory nerve endings to modulate long-term emotional and cognitive centers associated with the reward value of food 1120.
Furthermore, neuroimmune communication routes highlight how gut health impacts cognitive regulation of appetite. Elevated levels of bacteria such as Morganella and Alistipes are linked to lipopolysaccharide (LPS)-mediated activation of TLR4 receptors. This localized disruption increases endotoxemia, activating systemic inflammation via cytokines like IL-6 and TNF-$\alpha$, which traffic through visceral adipose tissue to the brain. This "gut-fat-brain axis" modulates glial homeostasis, ultimately influencing cognition and the psychological drive to eat 18.
Metabolic Adaptation and Weight Loss Resistance
A prevailing obstacle in the clinical management of obesity is the high prevalence of weight regain following successful caloric restriction. Historically attributed to behavioral relapse or a lack of willpower, contemporary clinical endocrinology categorizes weight regain as a profound, biologically driven physiological response known as metabolic adaptation or adaptive thermogenesis 212229.
Adaptive Thermogenesis Mechanisms
When an individual loses significant body mass, the body perceives the negative energy balance as an acute threat to survival. In response, energy expenditure drops precipitously. Metabolic adaptation occurs when this decline in resting metabolic rate (RMR) exceeds what would be mathematically predicted based solely on the loss of fat mass and fat-free mass 2324. Longitudinal research tracking participants from severe weight-loss regimens (e.g., The Biggest Loser cohorts) revealed that individuals expected to experience an RMR drop of 175 kcal/day actually suffered a decline of 664 kcal/day - a gap of nearly 489 kcal that persisted for up to six years post-intervention 2129.
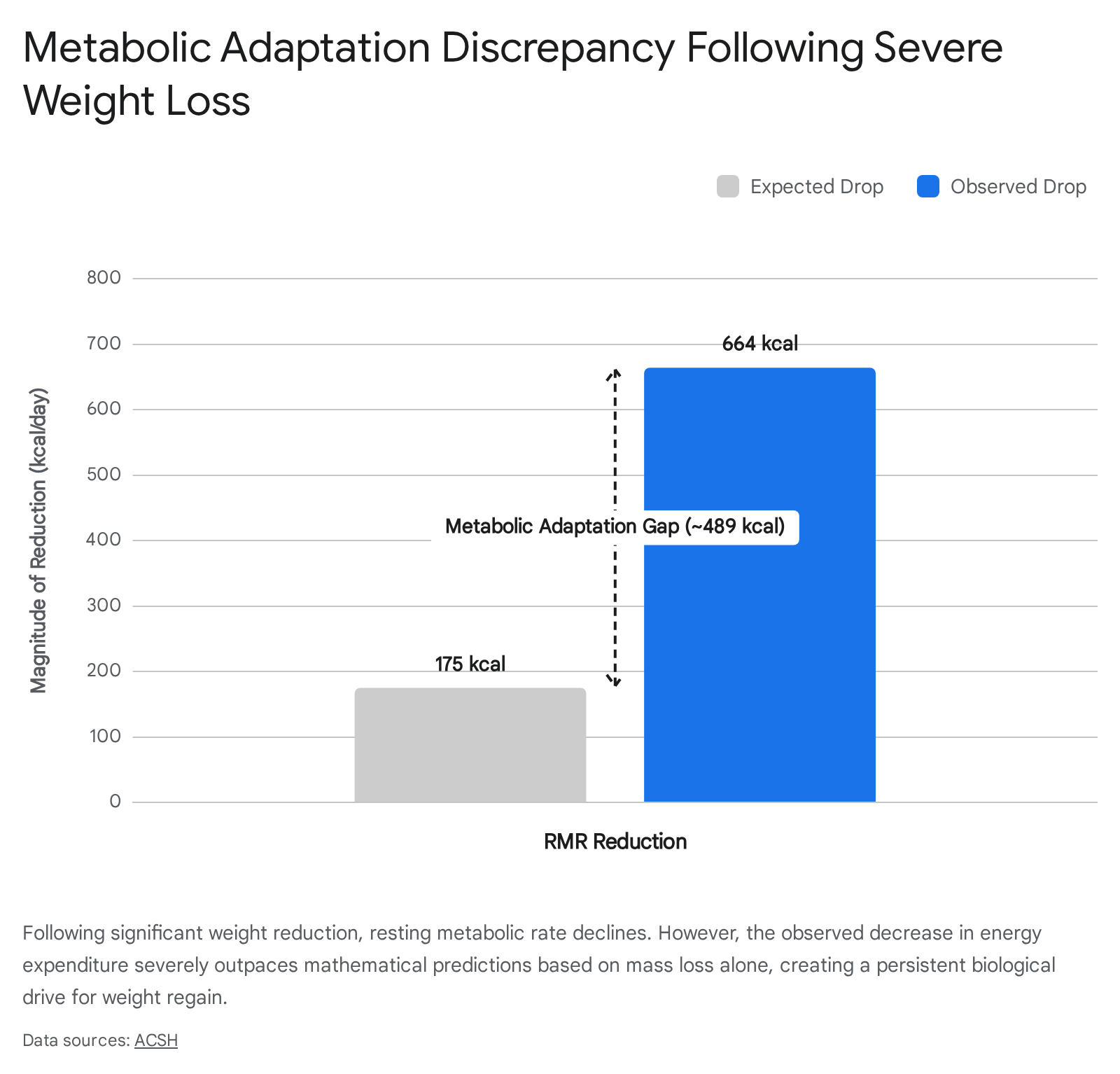
Clinical investigations from the University of Alabama at Birmingham (UAB) demonstrate that while metabolic adaptation does not directly force weight regain, it severely extends the time necessary to achieve weight loss goals and limits the total magnitude of fat mass reduction during low-energy diets 2325. Concurrently, weight loss triggers a persistent, coordinated shift in circulating appetite hormones. The adipose tissue secretes less leptin, removing the chronic stimulation of POMC satiety neurons. Simultaneously, gastrointestinal satiety hormones like CCK, PYY, and GLP-1 diminish, while the production of ghrelin spikes heavily, creating an intense neurological drive to eat that outlasts the diet itself 212226.
Mitochondrial Efficiency and Redox Metabolism
The persistence of metabolic adaptation fundamentally reframes obesity as a chronic, relapsing disease. Because the homeostatic mechanisms of the hypothalamus defend an elevated "set point," traditional dietary restriction faces continuous, escalating biological resistance. Recent 2025 breakthroughs in molecular endocrinology reveal that this resistance is tied deeply to cellular bioenergetics. Experimental work has demonstrated that mitochondrial electron transport functions as a dynamic redox regulator rather than a passive energy conduit. Imbalances in coenzyme Q and reverse electron transport are directly linked to hepatic steatosis, metabolic dysfunction, and cellular efficiency shifts that require less ATP to perform the same muscular work, effectively neutralizing the calorie-burning benefits of exercise 273528.
The success of modern incretin-based pharmacotherapies - such as tirzepatide (SURMOUNT trials) and the combination amylin/GLP-1 agonist CagriSema (REDEFINE trials) - lies in their ability to orchestrate a central nervous system bypass of these adaptive mechanisms 2429. By artificially maintaining high levels of simulated GLP-1, GIP, or amylin, these drugs force the continuous inhibition of AgRP neurons and the activation of POMC neurons despite the loss of fat mass. However, from a systems biology perspective, these agonists function as a metabolic bypass rather than a metabolic cure; they circumvent dysregulated appetite signaling but do not inherently restore fundamental mitochondrial efficiency or redox resilience, which explains the high rate of weight regain upon therapy discontinuation 28.
Disruption by Ultra-Processed Food Environments
The delicate neurobiological balance governing satiety is heavily disrupted by the modern food environment, characterized by the pervasive availability of ultra-processed foods (UPFs). A landmark 2025 Lancet Series extensively detailed how the rapid global integration of UPFs - defined by the NOVA classification as industrial formulations of cheap ingredients and cosmetic additives - is systematically undermining human metabolic health 303931.
Nutritional Displacement and Hyper-Palatability
UPFs are engineered for hyper-palatability, exploiting the brain's reward centers by offering highly concentrated doses of refined carbohydrates, free sugars, and saturated fats in tandem with flavor enhancers, emulsifiers, and texturizers 303141. This intentional formulation overrides the standard orosensory braking mechanisms governed by PRLH neurons; the food is digested and swallowed rapidly, outpacing the mechanical stretch signals of the stomach 641.
Furthermore, UPFs are characteristically low in fiber, protein, and intact food matrices. Because the structural matrix of the food is destroyed during industrial processing (e.g., extrusion, molding, pre-frying), digestion occurs artificially fast in the upper gastrointestinal tract 393141. Consequently, less chyme reaches the distal intestine to stimulate the L-cells, leading to an attenuated release of critical satiety hormones like GLP-1 and PYY 39. Controlled clinical feeding trials referenced in the 2025 Lancet analysis demonstrated that diets matching macronutrient profiles but differing in processing levels resulted in subjects unconsciously consuming approximately 500 surplus kilocalories per day when restricted to the UPF diet, resulting in an average weight gain of nearly one kilogram over just two weeks 393233.
Global Consumption Trends and Disease Incidence
The displacement of traditional, minimally processed diets by UPFs is a global phenomenon driven by massive corporate marketing and supply chain dominance 3034. The 2025 Lancet Series reviewed sales data from 93 countries and detailed an unprecedented structural shift in human dietary intake, where multinational corporations have aggressively lobbied against public health interventions to maintain market expansion 313536.
| Region / Country | Historical UPF Energy Share | Current (2024/2025) UPF Energy Share | Trend Description |
|---|---|---|---|
| United States & UK | > 50% | > 50% | Dominant dietary staple; modest recent increases due to prior saturation 313536. |
| Canada | 24.4% (80 years prior) | 54.9% | More than doubled over an 80-year horizon 31. |
| Spain | 11.0% (30 years prior) | 31.7% | Tripled over a three-decade span 3135. |
| Brazil & Mexico | ~10.0% (40 years prior) | ~23.0% | Doubled over four decades, indicating rapid market penetration 3135. |
| China | 3.5% | 10.4% | Tripled, though still a relatively minor overall share compared to the West 31. |
The epidemiological consensus links these high-UPF dietary patterns to significant increases in severe chronic disease. A systematic review of 104 long-term studies found that 92 showed higher risks for at least one chronic disease. Meta-analyses identified significant associations between high UPF intake and 12 distinct health conditions, including up to a 13% greater overall cancer risk, a 30% greater risk of ovarian cancer mortality, and three to four times the likelihood of requiring surgery for inflammatory bowel disease 3337. High consumption is also associated with up to a 28% higher risk of all-cause mortality during study follow-ups 33. While scientific debate continues regarding whether the harm is entirely driven by poor nutrient profiles or if industrial additives intrinsically disrupt gut barrier function and the microbiome, the overarching impact on satiety failure and global obesity rates is irrefutable 3933.