Chronic diphenhydramine use and dementia risk
Pharmacological Profile of Diphenhydramine
Diphenhydramine is a lipophilic, first-generation histamine H1-receptor antagonist that has been a mainstay of both prescription and over-the-counter pharmacotherapy since its introduction in the mid-twentieth century 123. While its primary clinical indications involve the management of allergic rhinitis, pruritus, and acute hypersensitivity reactions, its pronounced sedative properties have led to widespread, chronic utilization as a non-prescription sleep aid for transient and chronic insomnia 145. The pharmacological complexity of diphenhydramine arises from its lack of receptor specificity. In addition to antagonizing peripheral and central H1 receptors, the molecular structure of diphenhydramine confers a high binding affinity for muscarinic acetylcholine receptors 167.
This systemic muscarinic receptor antagonism is the biological foundation of the drug's anticholinergic adverse effect profile. Acute peripheral manifestations of this blockade routinely include xerostomia (dry mouth), mydriasis (pupillary dilation), blurred vision, diminished gastrointestinal motility leading to constipation, and urinary retention 18910. However, the critical pharmacokinetic characteristic distinguishing first-generation antihistamines from their second-generation counterparts is their high lipid solubility and low molecular weight, which facilitate rapid and extensive penetration across the blood-brain barrier 111213.
Within the central nervous system, acetylcholine operates as a ubiquitous excitatory neurotransmitter, modulating neural circuits intrinsic to arousal, attention, sensory processing, learning, and the consolidation of short-term memory into long-term storage 4141516. By competitively inhibiting central muscarinic receptors, diphenhydramine directly impedes cholinergic signal transduction. In a healthy adult, this blockade induces acute, theoretically reversible cognitive dulling, somnolence, and impaired psychomotor vigilance 121517. Clinical trials and simulated performance studies have demonstrated that standard therapeutic doses of diphenhydramine (e.g., 50 milligrams) can impair driving performance, choice reaction time, and working memory to a degree comparable to or exceeding the impairment observed at legal limits of blood alcohol concentration 5121518.
Age-Related Pharmacokinetic Alterations
The neurochemical impact of diphenhydramine is exponentially amplified in the geriatric population due to predictable, age-associated physiological shifts. As the human brain ages, there is a natural decline in cholinergic reserves, a reduction in muscarinic receptor density, and structural degradation of the basal forebrain cholinergic projection system 171920. Concurrently, the permeability of the blood-brain barrier increases with advanced age and microvascular disease, permitting higher intracerebral concentrations of lipophilic drugs 1719.
Furthermore, standard hepatic metabolism (primarily via the cytochrome P450 system) and renal clearance mechanisms deteriorate over time, extending the biological half-life of diphenhydramine 1417. In populations with end-stage renal disease undergoing hemodialysis, diphenhydramine presents a unique toxicological hazard. The molecule exhibits aggressive protein binding, often exceeding 95 percent albumin binding, which severely limits its dialytic clearance 21. Consequently, standard dosing regimens in these patients lead to rapid drug accumulation, prolonged systemic exposure, and a substantially heightened risk for severe central nervous system depression, profound delirium, and falls 2122. Because of this vulnerability, leading geriatric guidelines globally advise strict caution or absolute avoidance of diphenhydramine in older adults 81519.
Quantification of Anticholinergic Exposure
To robustly investigate the epidemiological relationship between long-term diphenhydramine exposure and irreversible neurodegenerative conditions, researchers required standardized methodologies capable of quantifying both the intrinsic potency of varying anticholinergic agents and the cumulative volume of patient exposure over a span of decades 192324. Because older adults frequently present with multimorbidity and subsequent polypharmacy - routinely combining over-the-counter antihistamines with prescription tricyclic antidepressants, urological antispasmodics, and antipsychotics - the analytical models must account for an aggregate "anticholinergic burden" 9252627.
Evolution of Anticholinergic Rating Scales
Over the past two decades, numerous clinical and pharmacological scales have been developed to stratify medications based on their in vitro muscarinic receptor affinity and their clinically observed propensity to trigger anticholinergic adverse events 192427. These tools are utilized extensively in both pharmacoepidemiology and frontline clinical decision support 2728.
| Anticholinergic Scale | Methodological Foundation | Diphenhydramine Classification | Primary Utility and Limitations |
|---|---|---|---|
| Anticholinergic Cognitive Burden (ACB) Scale | Systematic literature review, expert multidisciplinary consensus, and in vitro muscarinic receptor binding data. Scores range from 1 (possible) to 3 (definite central cognitive risk) 272930. | Score 2 or 3 (Definite central anticholinergic activity and known cognitive risk) 1031. | Most widely validated scale for forecasting dementia and cognitive decline. Limited by its failure to incorporate dose variation 242627. |
| Anticholinergic Risk Scale (ARS) | Expert consensus review focusing primarily on the prevalence of acute adverse clinical events. Ranks medications from 0 (low risk) to 3 (high risk) 3233. | Score 1 or 2 (Moderate to high risk of adverse events) 3234. | Highly predictive for acute peripheral events (falls, constipation, urinary retention) and delirium, though less precise for longitudinal cognitive decline 323335. |
| Drug Burden Index (DBI) | Continuous pharmacologic modeling combining cumulative prescribed dose relative to the recommended minimum effective clinical dose 192436. | Computed as a continuous, dose-dependent variable 3637. | Excellent for real-time functional impairment assessment in complex multimorbid patients, but computationally intensive for large historical datasets 243637. |
| Anticholinergic Medication Index (ACMI) | Machine learning-driven prognostic modeling applied to massive electronic health record (EHR) databases, indexing actual hospitalization rates 283839. | Classified as a high-burden indicator contributing to aggregate risk 3839. | Translates easily into automated EHR alerts. Externally validated in the UK Biobank for mortality and dementia risk stratification 283839. |
Despite the widespread adoption of these scales, significant heterogeneity remains. Different expert panels frequently assign divergent scores to the same molecule based on the specific clinical populations studied or the weighting of in vitro versus in vivo data 27. For example, certain atypical antipsychotics may receive a severe score of 3 on one scale and a mild score of 1 on another 27. Nevertheless, diphenhydramine is universally categorized as a high-potency, definite anticholinergic across virtually all established instruments 103140.
Standardization of Dose Metrics
Categorical rating scales identify which drugs carry risk, but assessing chronic exposure requires the integration of dispensing data over time. Researchers predominantly utilize the Total Standardized Daily Dose (TSDD) or the cumulative Defined Daily Dose (cDDD) 2341.
The TSDD methodology involves extracting the total milligrams of a target medication dispensed over a predefined exposure window and dividing that sum by the minimum effective daily maintenance dose recommended for a geriatric population 172342. This mathematical conversion standardizes vastly different chemical entities into a single, uniform exposure metric 2343. For example, accumulating a TSDD of 1095 indicates that an individual has consumed the equivalent of the minimum effective daily dose of an anticholinergic drug every single day for three years 234344. In long-term cohort studies, total cumulative exposure is typically categorized into defined thresholds: non-use (0 TSDD), low exposure (1 - 90 TSDD), moderate exposure (91 - 365 TSDD), high exposure (366 - 1095 TSDD), and severe exposure (>1095 TSDD) 172325.
Epidemiological Evidence for Dementia Associations
The hypothesis that chronic utilization of diphenhydramine and structurally related anticholinergic agents directly contributes to the pathogenesis of incident dementia relies heavily on an expanding corpus of robust, prospective cohort studies and nested case-control analyses 17454648. These pharmacoepidemiological investigations consistently report a statistically significant, dose-dependent escalation in dementia risk corresponding with a higher cumulative anticholinergic burden 174647.
Foundational Cohort and Case-Control Studies
The paradigm surrounding anticholinergic safety shifted significantly following the publication of a landmark 2015 prospective cohort study by Gray et al. in JAMA Internal Medicine 5174849. Utilizing data from the Adult Changes in Thought (ACT) study within a Seattle-based integrated healthcare system, researchers tracked 3,434 older adults who were free of dementia at baseline for a mean follow-up duration of 7.3 years 1749. During the observation period, 797 participants developed incident dementia, with the vast majority presenting with Alzheimer's disease pathology 1749.
The ACT study identified a striking 10-year cumulative dose-response relationship. Participants exhibiting the highest cumulative anticholinergic exposure - defined as greater than 1095 TSDDs, driven primarily by first-generation antihistamines like diphenhydramine, tricyclic antidepressants, and bladder antimuscarinics - demonstrated a 54 percent increased relative risk for incident dementia (adjusted hazard ratio [aHR] 1.54; 95% Confidence Interval [CI] 1.21-1.96) when compared to non-users 172549. Most alarmingly, the elevated risk profile persisted even among individuals who had ceased taking anticholinergic medications years prior to their cognitive assessment, directly challenging the long-held medical consensus that anticholinergic cognitive impairment is entirely transient and fully reversible upon drug discontinuation 4850.
These findings were subsequently validated by massive international database analyses. A 2019 nested case-control study by Coupland et al. analyzed 58,769 patients diagnosed with dementia against 225,574 matched controls within the United Kingdom's general practice registries 515253. The researchers confirmed the dose-dependent risk across multiple drug classes, finding significant associations for exposure occurring 1 to 11 years prior to diagnosis 2553. Similarly, a 2018 British case-control study by Richardson et al., comprising 40,770 dementia cases and 283,933 controls, reported a generalized elevated odds ratio (OR 1.11; 95% CI 1.08-1.14) for prior anticholinergic exposure, with a distinct exposure-dependent gradient 255054.
Exposure Dose-Response Gradients and Subtype Disparities
The consistency of the dose-response gradient is one of the strongest epidemiological arguments supporting a potential causal link. A 2025 nationwide Swedish case-control study led by Zhu et al. evaluated 199,526 individuals with incident dementia and an equivalent control group, generating highly granular exposure data 47. The researchers demonstrated that the risk of all-cause dementia rose strictly in tandem with cumulative exposure to strong anticholinergics 47.
| Cumulative Exposure Tier (Defined Daily Doses) | Adjusted Odds Ratio (aOR) for Incident Dementia | 95% Confidence Interval & Statistical Significance |
|---|---|---|
| Low Exposure (1-89 DDDs) | 1.10 | P < .001 47 |
| Moderate Exposure (90-364 DDDs) | 1.39 | P < .001 47 |
| High Exposure (365-1094 DDDs) | 1.57 | P < .001 47 |
| Severe Exposure (≥ 1095 DDDs) | 1.66 | P < .001 47 |
While the link to generalized dementia is clear, the associations across specific neurodegenerative subtypes present intriguing complexities. While Gray et al. reported strong correlations with Alzheimer's disease, other vast cohorts indicate that the anticholinergic risk is noticeably more pronounced for vascular dementia and Lewy Body Dementia than for classical Alzheimer's disease 2547. Furthermore, the Swedish cohort noted that the risk amplification was strongest in younger individuals experiencing early-onset cognitive decline and particularly distinct among men 47. These subtype disparities suggest that the mechanism of injury may heavily involve neurovascular components or specific dopaminergic/cholinergic network disruptions rather than universally accelerating widespread cortical amyloidosis 2547.
Comparative Risk Profiles of Antihistamine Generations
The anticholinergic hypothesis is further refined when analyzing specific medication classes. First-generation antihistamines (FGAs) like diphenhydramine, chlorpheniramine, and doxylamine are structurally engineered to cross the blood-brain barrier 11155. Conversely, second-generation antihistamines (SGAs) such as loratadine, cetirizine, and fexofenadine possess bulky, lipophobic side chains that drastically limit their central nervous system penetration, rendering them clinically non-sedating under normal physiological conditions 111245.
If central muscarinic blockade is the primary driver of incident dementia, the epidemiological data should demonstrate a stark divergence in long-term risk between FGAs and SGAs. This hypothesis was rigorously tested in a 2024 retrospective cohort study by Su et al., published in The Journal of Allergy and Clinical Immunology: In Practice 114158. Utilizing Taiwan's National Health Insurance Research Database, the researchers tracked 714,052 patients diagnosed with new-onset allergic rhinitis over a six-year period 11. The cohort was stratified into non-users, FGA users, and SGA users, with exposure quantified via cumulative defined daily doses 1141.
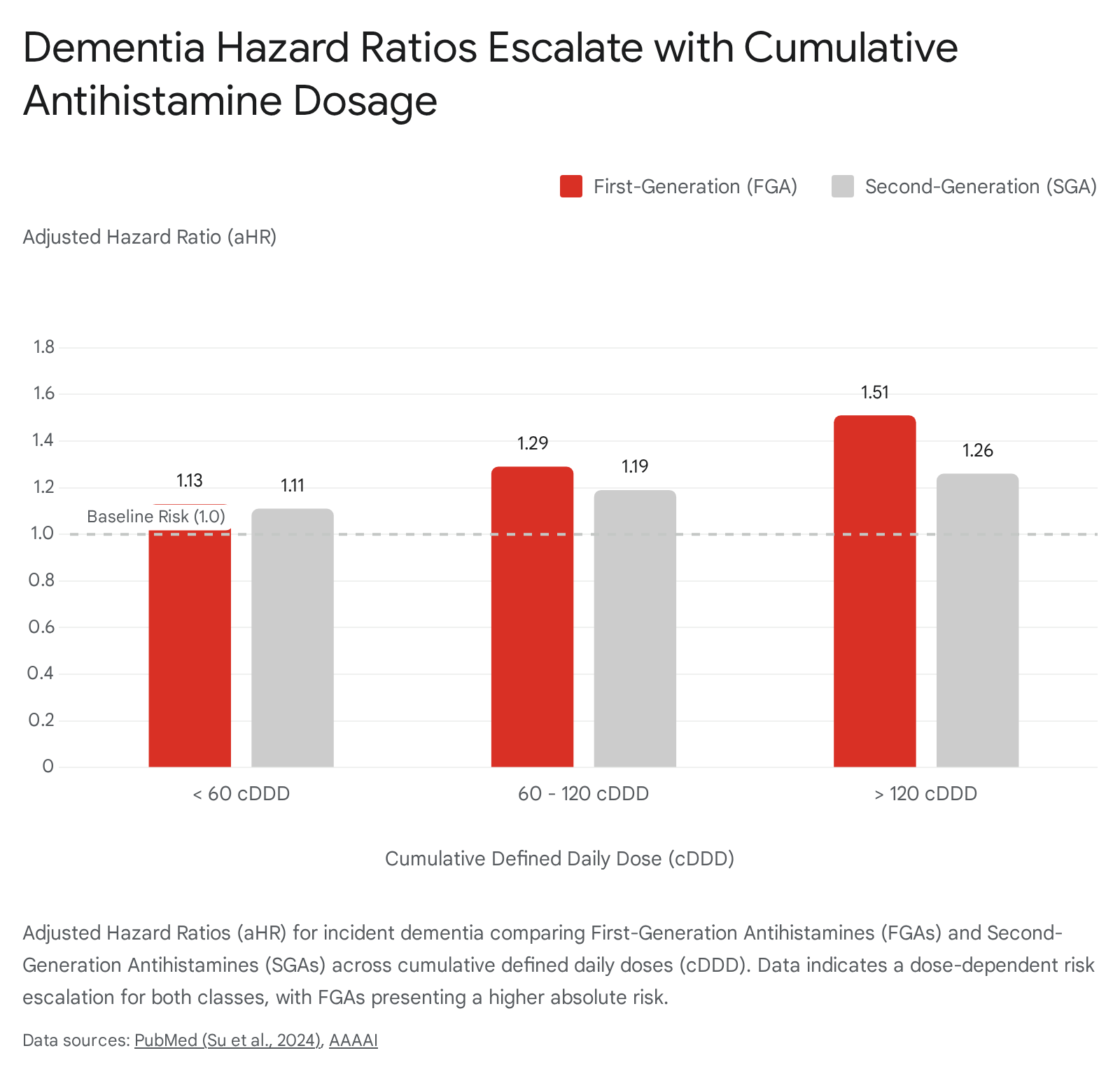
The Taiwanese cohort definitively established that first-generation antihistamines pose a significantly higher, dose-dependent risk for dementia compared to their second-generation equivalents 1141. At exposure levels exceeding 120 cDDD, FGA users exhibited an adjusted hazard ratio of 1.51, reflecting a steep escalation in risk profile 1141.
However, the findings also revealed a critical nuance: second-generation antihistamines were not entirely devoid of risk. High cumulative exposure to SGAs (>120 cDDD) resulted in an adjusted hazard ratio of 1.26 1141. The presence of measurable cognitive risk associated with SGAs suggests several possibilities. Firstly, while SGAs are lipophobic, massive accumulation over long durations may still result in subtle blood-brain barrier penetration, particularly if the barrier is compromised by advancing age or systemic disease 1217. Alternatively, conditions characterized by chronic systemic inflammation or hypoxia - such as severe, uncontrolled allergic rhinitis - may themselves amplify cognitive deficits, compounding the marginal central inhibitory effects of the medications 12. Despite these marginal risks, clinical consensus remains clear: second-generation antihistamines demonstrate a vastly superior neurological safety profile compared to the severe central impact of first-generation agents 132145.
The Protopathic Bias and Reverse Causality Dilemma
While large-scale observational data robustly support an association between chronic diphenhydramine utilization and subsequent dementia, asserting a direct, causal sequence of events remains highly controversial. The primary impediment to establishing causality is a persistent epidemiological phenomenon known as protopathic bias, frequently referred to in this context as reverse causality 172550.
Protopathic bias manifests when a pharmacological agent is prescribed or self-administered to mitigate the early, unrecognizable symptoms of a disease long before that disease progresses to the point of clinical diagnosis 175054. The neurodegenerative processes defining Alzheimer's disease, Lewy Body Dementia, and other related disorders do not begin on the day of formal diagnosis; rather, cortical amyloidosis, tau protein aggregation, and cholinergic neuronal death initiate silently up to twenty years prior to the onset of frank memory failure 175356.
During this extensive preclinical or prodromal phase, patients routinely experience insidious, non-specific physiological and psychological disruptions. Among the most prevalent prodromal symptoms are severe sleep architecture fragmentation, chronic insomnia, escalating anxiety, and mild depressive states 172050. Because diphenhydramine is an inexpensive, over-the-counter medication explicitly marketed for its sedative properties (as the primary active ingredient in "PM" analgesics and standalone sleep aids), older adults suffering from prodromal insomnia frequently self-medicate with high cumulative doses over several years 4505157.
Consequently, the elevated incidence of dementia observed in cohorts of chronic diphenhydramine users may not constitute evidence that the drug triggered the neurodegeneration. Instead, it strongly suggests that the underlying, subclinical neurodegeneration induced sleep disturbances, which subsequently drove the patient to consume the drug 1750. In this scenario, diphenhydramine use serves as a pharmacological marker of impending dementia rather than its biological origin.
REM Sleep Behavior Disorder as a Confounding Catalyst
The biological reality of the reverse causality hypothesis is powerfully illuminated by specific, highly predictive sleep pathologies. Rapid-Eye-Movement (REM) Sleep Behavior Disorder (RBD) is a parasomnia in which the brain fails to induce the normal skeletal muscle atonia required during REM sleep 565859. As a result, patients physically, and often violently, enact their dreams 5658.
Extensive neurobiological research over the last decade has cemented RBD not merely as a standalone sleep disorder, but as a definitive prodromal manifestation of underlying alpha-synucleinopathies - specifically Parkinson's disease, Lewy Body Dementia, and multiple system atrophy 56596061. Autopsy studies confirm that up to 98 percent of individuals with polysomnogram-confirmed RBD exhibit synucleinopathy pathology, and roughly 80 percent of patients ultimately diagnosed with Lewy Body Dementia report a prior history of RBD, often preceding their cognitive decline by over a decade 5660.
The structural degradation of brain stem regions controlling motor movements during sleep, combined with general overactivation of the ascending reticular activating system, induces profound insomnia and sleep cycle disruption in these patients 58. Desperate for rest, these individuals frequently seek out potent sedative-hypnotics, including highly anticholinergic antihistamines 5859.
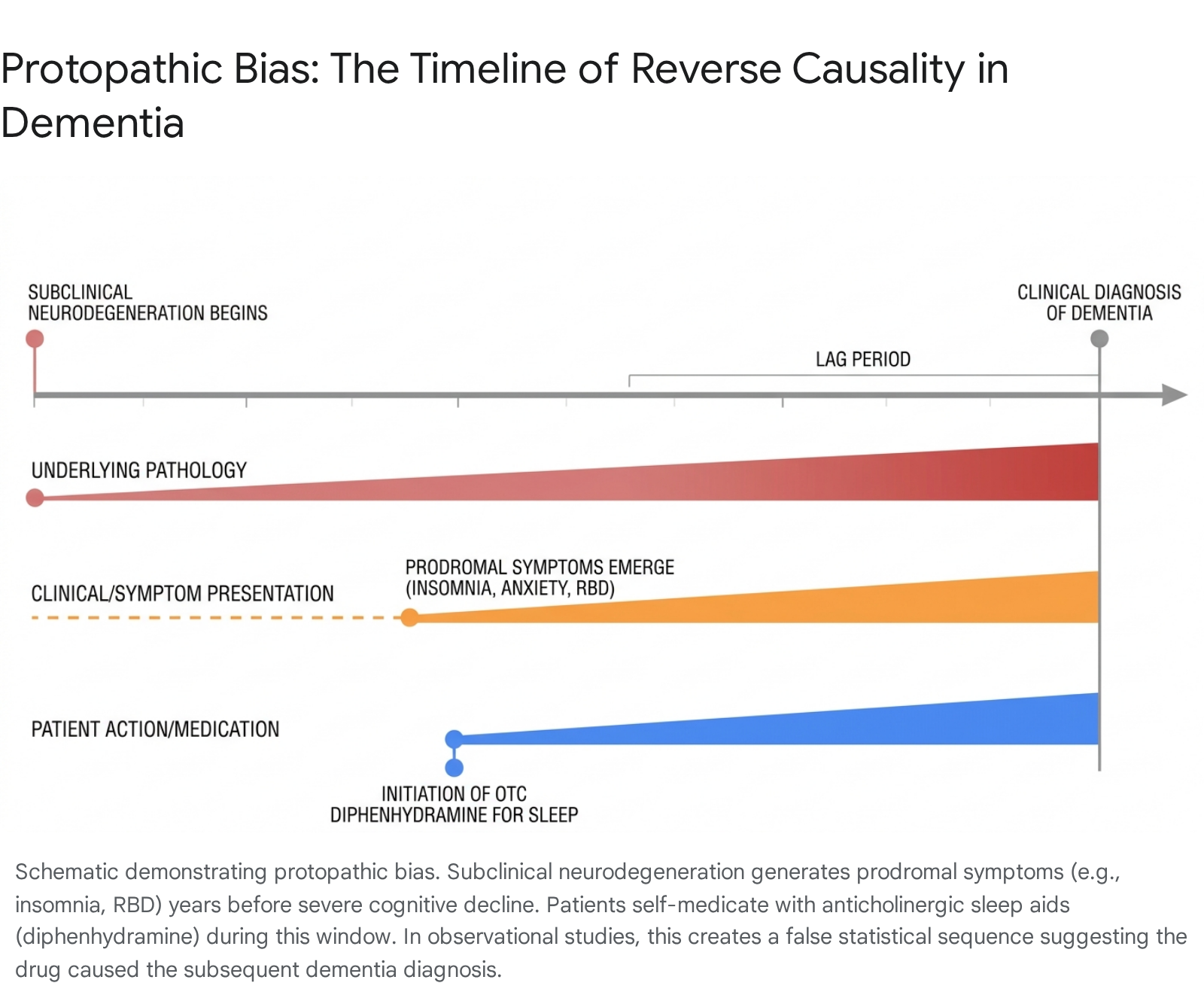
Therefore, the statistical correlation between anticholinergic use and subsequent dementia diagnosis in these specific cohorts is almost certainly heavily polluted by reverse causality originating from the prodromal parasomnias 5859.
Methodological Mitigations and Lag Period Implementation
To isolate potential direct neurotoxic causality from protopathic bias, high-quality pharmacoepidemiological studies construct temporal statistical "lag periods" 25425462. This methodology systematically excludes any medication exposure that occurred within a defined window immediately preceding the formal dementia diagnosis, operating on the assumption that recent prescriptions were likely addressing prodromal symptoms rather than initiating the disease process 4254.
The length of the applied lag period varies considerably across studies, directly impacting their conclusions. Gray et al. utilized a conservative 12-month lag period, acknowledging that while it excludes immediate pre-diagnosis exposure, it may fail to account for prodromal states lasting a decade 549. A more rigorous approach by Richardson et al. analyzed a massive UK general practice cohort by completely excluding all drug exposures within the four years preceding the index date, attempting to sever the reverse causality link entirely 54. The most exhaustive models, such as the one employed by Coupland et al., tested multiple stratified exposure windows, analyzing drug use recorded 1 to 11 years, 3 to 13 years, and up to 5 to 20 years before the identification of dementia 2553.
Crucially, even when implementing aggressive lag periods extending up to two decades prior to diagnosis, several of these massive studies continued to identify a statistically significant, dose-dependent escalation in dementia risk correlated with cumulative anticholinergic burden 255354. The persistence of this risk across expansive temporal voids suggests that while protopathic bias is an undeniable and powerful confounder, it may not exclusively account for the entirety of the observed cognitive decline, leaving open the possibility of a direct, neurotoxic mechanism 5354.
Critiques of the Causal Hypothesis
Despite the implementation of lag periods, elements of the causal hypothesis remain highly contested. Critics of the direct neurotoxicity model, notably articulated in psychiatric literature, point to profound inconsistencies within the observational data 25. If systemic anticholinergic activity acts as a universal neurotoxin, the risk elevation should theoretically manifest uniformly across all pharmacological categories sharing that mechanism of action 25.
However, the data frequently reveals extreme drug class disparities. Multiple massive cohorts have observed that the increased dementia risk is heavily driven by select categories - specifically, high-potency tricyclic antidepressants, antipsychotics, and urological antispasmodics - while other highly anticholinergic classes, such as gastrointestinal antispasmodics, fail to demonstrate any clear longitudinal association with cognitive decline 2554. Furthermore, the statistical finding that an exposure equivalent to merely 1 to 90 total standardized daily doses could precipitate a measurable increase in dementia risk up to 20 years later stretches biological plausibility 25. A compound capable of inflicting such widespread, permanent cortical damage from brief, distant exposure would represent a neurotoxin of unprecedented severity, an effect disproportionate to the clinical profile of over-the-counter antihistamines 25. These anomalies suggest that "confounding by indication" - where the underlying physiological condition necessitating the prescription (e.g., severe mood disorders, chronic neurogenic bladder) is the true driver of the dementia risk - remains a pervasive, unresolved flaw in the epidemiological framework 2562.
Putative Biological Mechanisms of Neurotoxicity
If chronic exposure to lipophilic anticholinergic agents like diphenhydramine does indeed enact a direct, causal influence on the acceleration or initiation of neurodegenerative disease, the underlying pathophysiology must extend beyond the transient, reversible receptor blockade traditionally recognized 51748.
Cholinergic Depletion and Cellular Stress
The foundational mechanism of Alzheimer's disease involves the early and progressive degeneration of cholinergic neurons originating in the basal forebrain 4863. In a healthy, aging brain, physiological compensatory mechanisms attempt to maintain cognitive equilibrium despite gradual neuronal attrition 17. Chronic, high-dose administration of central muscarinic antagonists introduces a profound artificial stressor to an already vulnerable network 1417. Continuous pharmacological blockade of the surviving muscarinic receptors may force the remaining, stressed cholinergic neurons to upregulate neurotransmitter synthesis or alter receptor density, thereby accelerating cellular exhaustion and premature apoptosis 121417. This sustained interference theoretically compromises the brain's cognitive reserve, precipitating an earlier clinical transition from manageable mild cognitive impairment into profound, irreversible dementia 121417.
Neuroanatomical Atrophy and Biomarker Evidence
Emerging neuroimaging and biomarker research provides tentative structural support for the neurotoxicity hypothesis. Subset analyses investigating patients with high cumulative anticholinergic burdens have identified distinct patterns of cerebral atrophy 636465. High-resolution magnetic resonance imaging (MRI) studies comparing chronic anticholinergic users to matched non-users have reported correlations with reduced total cortical volume, enlarged ventricular spaces, and specific, localized cross-sectional atrophy within the basal forebrain, temporal lobes, and the hippocampus - regions critical for memory encoding and early targets of Alzheimer's pathology 636465.
Furthermore, biochemical analysis exploring neurofilament light chain (NfL) - a highly sensitive protein biomarker released into blood plasma upon axonal damage and neurodegeneration - has revealed concerning synergies. A 2025 prospective cohort analysis from the Shanghai Aging Study, tracking 1,529 dementia-free adults, discovered that patients presenting with both elevated baseline NfL levels and a history of anticholinergic exposure faced an exponentially higher cumulative risk of incident dementia than those with elevated NfL alone 66. This interaction suggests that anticholinergic drugs may act as a catalytic force, aggressively accelerating pre-existing subclinical neurodegeneration 66. Additionally, recent advances utilizing Mendelian randomization - a robust statistical technique that leverages genetic variants as instrumental variables to bypass environmental confounders - have indicated that systemic antihistamines present a probable causal, negative effect on overall cognitive performance, further undermining the argument that the risk is entirely artifactual 67.
Clinical Guidelines and Deprescribing Strategies
The intense scientific debate regarding absolute causality versus protopathic bias ultimately yields to a unified, unambiguous clinical consensus: the chronic, unmonitored use of diphenhydramine and structurally similar first-generation antihistamines presents an unacceptable hazard to the geriatric population and must be aggressively avoided 8485171.
The American Geriatrics Society (AGS) Beers Criteria explicitly designates diphenhydramine as a highly inappropriate medication for adults aged 65 and older 815. This rigid classification is driven not solely by the debated long-term dementia risks, but by the immediate, pervasive incidence of acute cognitive impairment, severe delirium, orthostatic hypotension leading to catastrophic falls, and compounding peripheral toxicities resulting from age-related pharmacokinetic delays 8151955.
Alternative Interventions for Insomnia and Allergies
The ubiquitous availability of diphenhydramine within over-the-counter nighttime analgesics and allergy formulations requires aggressive patient education and proactive deprescribing efforts by frontline clinicians 577168.
For the management of allergic rhinitis, international clinical guidelines universally reject the first-line use of diphenhydramine in older adults 182145. Therapy must pivot to second-generation H1 antagonists - such as cetirizine, loratadine, or fexofenadine - which offer equivalent or superior peripheral symptom control while largely circumventing the blood-brain barrier, thereby preserving cognitive integrity 132145.
The utilization of diphenhydramine as a long-term intervention for chronic insomnia is considered poor medical practice across all age demographics 686970. The American Academy of Sleep Medicine strongly recommends against the use of over-the-counter antihistamines for sleep maintenance due to rapid tolerance development, daytime somnolence, and the severe anticholinergic side effect profile 7071. Optimal management of sleep architecture disruption requires a multifaceted approach prioritizing Cognitive Behavioral Therapy for Insomnia (CBT-I) 46869. When non-pharmacological interventions fail, clinicians must bypass anticholinergic agents in favor of modern, targeted pharmacotherapies, such as dual orexin receptor antagonists (DORAs like suvorexant), low-dose melatonin, or the strictly monitored, brief use of short-acting non-benzodiazepine hypnotics, continually balancing the immediate necessity of sleep against the long-term imperative of neurological preservation 59686971.