Cholinergic Regulation of Attention and Memory in Alzheimer's Disease
The cholinergic system serves as a foundational neuromodulatory network in the mammalian central nervous system, governing state-dependent processes ranging from arousal and vigilance to complex cognitive functions such as selective attention, learning, and episodic memory consolidation. Originating primarily from a collection of discrete nuclei in the basal forebrain, cholinergic projections blanket the cerebral cortex and hippocampus. In the context of Alzheimer disease (AD), the degradation of this system was originally conceptualized as a late-stage hallmark responsible for profound cognitive decline. However, recent evidence reframes cholinergic dysregulation as a highly dynamic, biphasic process intertwined with amyloid and tau pathologies, genetic vulnerabilities, and demographic factors 123. Understanding the precise anatomical, molecular, and pharmacological dimensions of the cholinergic system is critical for interpreting the current landscape of AD therapeutics, which is pivoting from legacy acetylcholinesterase inhibitors toward highly selective muscarinic receptor agonists.
Anatomical Organization of the Basal Forebrain Cholinergic System
The central source of acetylcholine (ACh) for the cortical and limbic regions is the basal forebrain cholinergic system (BFCS). The BFCS does not operate as a single homogeneous entity; rather, it is structurally partitioned into distinct sub-nuclei characterized by highly specialized topographical projection patterns that dictate functional specificity across the cerebral mantle.
Septohippocampal Pathways and Mnemonic Processing
The neurons of the BFCS are classified into four major cholinergic cell groups, designated Ch1 through Ch4, based on their anatomical location and target projection areas 145.
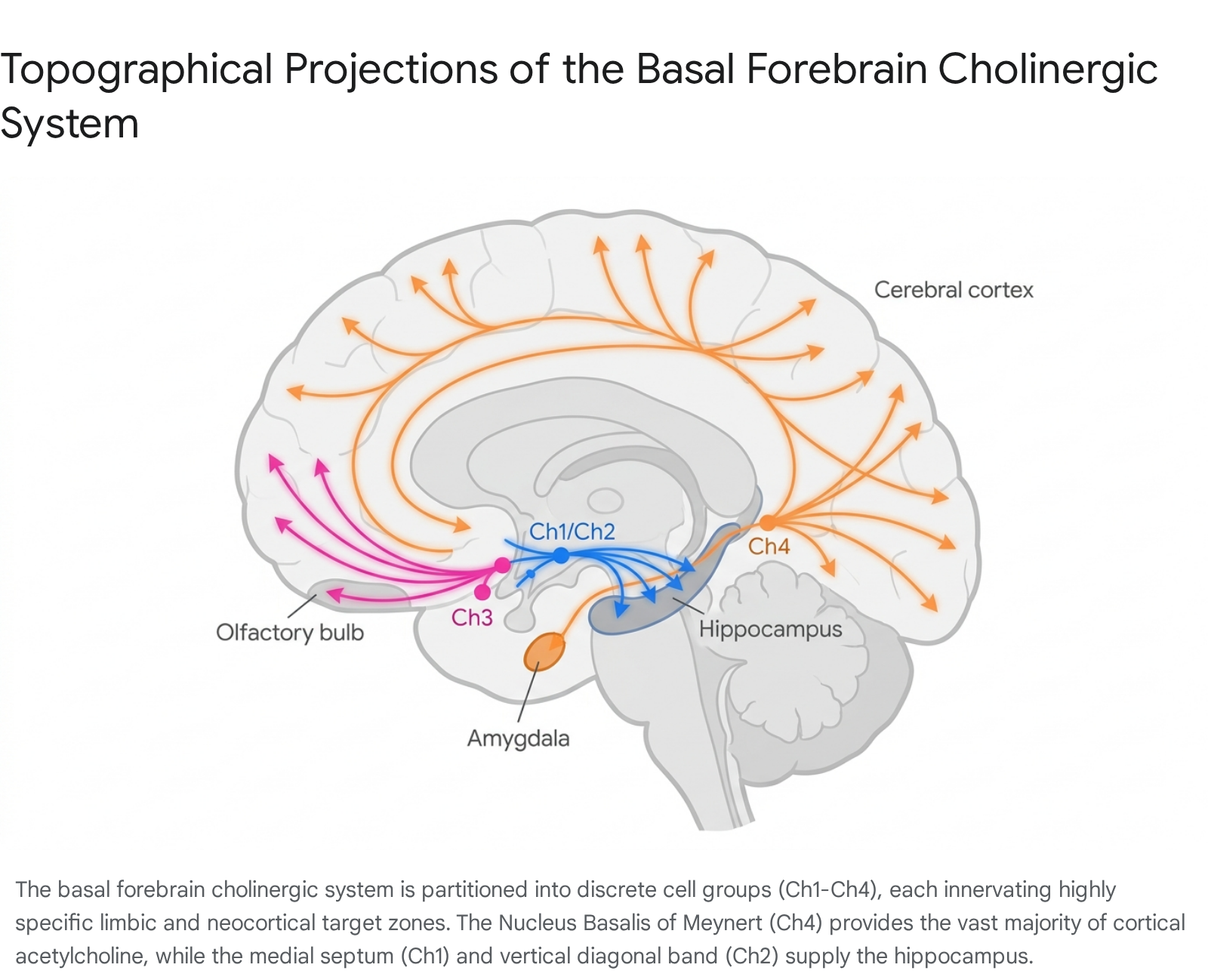
The Ch1 sector corresponds to the medial septal nucleus, while Ch2 refers to the vertical limb of the diagonal band of Broca. Together, these nuclei provide the predominant cholinergic innervation to the hippocampus and entorhinal cortex via the septohippocampal pathway 16.
The projections from Ch1 and Ch2 target specific hippocampal subfields, including the cornu ammonis sectors (CA1, CA2, CA3), the dentate gyrus, and the subiculum 78. The CA1 field, which receives massive laminar-specific information, integrates these cholinergic inputs to support spatial orientation, learning, and episodic memory. Notably, the CA1 subfield maintains reciprocal projections back to the medial septal nucleus and the vertical limb of the diagonal band, establishing a continuous feedback loop that modulates hippocampal theta oscillations necessary for the encoding of contextual events 910. Atrophy within the Ch1 and Ch2 nuclei, and the consequent loss of septohippocampal connectivity, correlates directly with the severe visuospatial memory impairments observed in the early clinical stages of neurodegenerative dementias 811.
The Nucleus Basalis of Meynert and Cortical Innervation
The Ch4 sector, anatomically synonymous with the nucleus basalis of Meynert (NBM), represents the largest contiguous cluster of cholinergic neurons in the human brain, containing approximately 210,000 cholinergic cells per hemisphere 10. The NBM provides the principal source of ACh to the entire cerebral cortex and the amygdala 412.
The Ch4 complex exhibits a highly organized internal topography that dictates its cortical targets. The anteromedial subfield (Ch4am) directs projections to medial cortical areas, including the cingulate gyrus. The anterolateral subfield (Ch4al) innervates frontoparietal cortices, opercular regions, and the amygdaloid nuclei. The intermediate subfield (Ch4i) targets the laterodorsal frontoparietal, peristriate, and midtemporal cortices, whereas the posterior subfield (Ch4p) predominantly serves the superior temporal and temporopolar regions 57. This discrete parcellation implies that localized neurodegeneration within specific NBM subfields yields highly specific cognitive and behavioral deficits. Quantitative volumetric analyses indicate that atrophy in the Ch4p and Ch4a-i subregions is detectable during subjective cognitive decline (SCD) and correlates strongly with emerging deficits in language and executive function 7.
Cytoarchitectonic Gradients and Limbic Vulnerability
Cholinergic innervation across the human cerebral cortex is not uniform; it is organized by a density gradient that aligns closely with hierarchies of information processing. Primary sensory and motor cortices exhibit intermediate to light cholinergic innervation, whereas unimodal and heteromodal association areas receive denser inputs 1314. The heaviest cholinergic innervation is found within paralimbic (e.g., insular, parahippocampal) and core limbic structures, including the hippocampus and the amygdala 414.
These limbic and paralimbic cortices serve a dual role: they are the primary recipients of Ch4 innervation and the exclusive sources of reciprocal feedback projections returning to the nucleus basalis 415. Because the NBM is deeply enmeshed within limbic circuitry, it is positioned to modulate the encoding and memorability of sensory experiences. However, this dense interconnectivity with limbic structures also renders the NBM anatomically contiguous with the earliest sites of neurofibrillary tau degeneration, establishing a structural basis for its profound vulnerability to the progression of Alzheimer pathology 45.
Molecular Architecture of Cholinergic Receptors
The effects of ACh on postsynaptic neurons are mediated by two distinct classes of receptors: G-protein-coupled muscarinic acetylcholine receptors (mAChRs) and ligand-gated ionotropic nicotinic acetylcholine receptors (nAChRs).
Muscarinic Receptor Subtypes and Signal Transduction
Muscarinic receptors consist of five subtypes (M1 - M5), divided into two distinct functional families based on their intracellular signaling cascades 1617.
The M1-like family includes the M1, M3, and M5 receptors, which are coupled to Gq/11 stimulatory proteins. Activation of these receptors stimulates phospholipase C, initiating the phosphatidylinositol trisphosphate cascade, which ultimately mobilizes intracellular calcium 16. In the central nervous system, M1 receptors are the most abundant mAChR subtype, heavily concentrated in the cerebral cortex and hippocampus. Postsynaptically, M1 receptors mediate sustained neuronal excitation and facilitate synaptic plasticity by potentiating N-methyl-D-aspartate (NMDA) receptor currents, making them indispensable for short-term memory and long-term cognitive modulation 161718.
The M2-like family comprises the M2 and M4 receptors, which are coupled to Gi/o inhibitory proteins. Their activation inhibits adenylyl cyclase, significantly reducing intracellular cyclic AMP (cAMP) levels 1617. M2 receptors act predominantly as presynaptic autoreceptors that inhibit the further release of ACh, providing a crucial negative feedback loop to regulate local cholinergic tone 1819. Decreased cAMP levels resulting from excessive M2 activity correlate with impaired synaptic plasticity and are linked to amnestic features in neurodegenerative models 16. M4 receptors, highly expressed in the striatum and moderately in the hippocampus and cortex, play a vital role in modulating dopaminergic release and have become a primary target for alleviating psychiatric and psychotic symptoms 1820.
Nicotinic Receptor Subtypes and Kinetic Properties
In the human brain, the most prominent nAChR subtypes are the homomeric α7 and the heteromeric α4β2 receptors 1621. The α7 subtype is heavily implicated in working memory and is uniquely concentrated in regions like the hippocampus and selected cortical layers, where it responds rapidly to cholinergic input and modulates the release of excitatory neurotransmitters. The α4β2 subtype is broadly distributed and is generally associated with the modulation of sustained attention and vigilance 2122.
Recent structural and pharmacological characterizations have expanded the understanding of nicotinic receptor complexes. While α7 receptors frequently assemble as homomers, affinity purification studies from surgically excised human temporal cortex demonstrate that α7 subunits also form functional heteromeric complexes with β2 subunits (α7β2 nAChRs) 23. These human α7β2 heteromers exhibit marked pharmacological and kinetic differences compared to α7 homomers, specifically displaying significantly slower rise and decay phases in response to cholinergic agonists 23. This kinetic distinction provides a mechanism for varied temporal integration of cholinergic signals in the cortex.
Laminar Specificity and the Signal-to-Noise Ratio
The distribution of muscarinic and nicotinic receptors is highly stratified across cortical layers, facilitating precise regulation of information flow within microcircuits.
| Receptor Subtype | Functional Family | Intracellular Coupling | Primary Cortical Lamination | Neurocognitive and Clinical Role |
|---|---|---|---|---|
| M1 | Muscarinic | Gq/11 (Stimulatory) | Supragranular (Layers II/III) | Promotes synaptic plasticity, episodic memory, and non-amyloidogenic APP processing. |
| M2 | Muscarinic | Gi/o (Inhibitory) | Granular/Infragranular (IV/V) | Presynaptic autoreceptor limiting ACh release; filters ascending sensory noise. |
| M3 | Muscarinic | Gq/11 (Stimulatory) | Sparsely distributed | Associated with sleep architecture and metabolic regulation. |
| M4 | Muscarinic | Gi/o (Inhibitory) | Striatum, Cortex, Hippocampus | Modulates dopamine release; primary target for mitigating psychosis and agitation. |
| M5 | Muscarinic | Gq/11 (Stimulatory) | Sparsely distributed | Regulates cerebral blood flow and vascular integrity. |
| α7 | Nicotinic | Ionotropic (Ligand-gated) | Supragranular, Hippocampus | Modulates working memory and rapid excitatory transmission. |
| α4β2 | Nicotinic | Ionotropic (Ligand-gated) | Granular/Infragranular (IV/V) | Modulates sustained attention and vigilant state maintenance. |
In the human neocortex, M1 receptors and α7 nAChRs achieve their highest densities within the supragranular layers (Layers II and III) 1924. Because these layers are the primary source of cortico-cortical efferent projections, M1 activation predominantly amplifies the processing and binding of high-order associative information 19. Conversely, M2 receptors and α4β2 nicotinic receptors are preferentially localized to the granular (Layer IV) and infragranular (Layer V) strata 192425. By inhibiting presynaptic transmitter release in Layer IV - the principal termination zone for ascending thalamocortical afferents - M2 receptors act to filter baseline sensory input. Together, this interlaminar distribution allows the cholinergic system to dynamically enhance the "signal-to-noise" ratio in cortical processing: damping bottom-up sensory noise via infragranular M2/α4β2 receptors while simultaneously boosting lateral, top-down cognitive signals via supragranular M1/α7 receptors 1217.
Cholinergic Dysregulation in Alzheimer Disease Pathogenesis
The classical "Cholinergic Hypothesis" of AD initially posited that a late-stage degeneration of cholinergic neurons in the basal forebrain yields a profound deficit in ACh, thereby driving the hallmark cognitive deficits of the disease 13. However, contemporary longitudinal data indicates that cholinergic dysfunction is not merely a late-stage consequence but rather an active, dynamic participant in the early cascade of AD pathophysiology.
Biphasic Disease Evolution and Early Hyperactivity
In the earliest stages of AD - often preceding objective cognitive decline - cholinergic neurons exhibit pathological overactivity rather than functional depression 2. High levels of amyloid precursor protein (APP) and low concentrations of soluble, oligomeric amyloid-beta (Aβ) actively stimulate cholinergic terminals. Aβ oligomers bind to p75 neurotrophin receptors (p75NTR) enriched on cholinergic synapses, which reduces specific potassium currents (such as calcium-activated and delayed rectifier currents) 2. This restriction of potassium efflux elevates extracellular potassium levels, driving the cholinergic terminal into a state of severe hyperexcitability.
Consequently, early AD is characterized by increased ACh release and neuronal hyperactivity, particularly driven by Ch1/Ch2 septohippocampal projections 23. This hyperexcitability manifests functionally as interictal spikes (IIS) occurring prominently in the dentate gyrus during sleep. These cholinergic-driven spikes disrupt the physiological sleep architecture required for the glymphatic clearance of neurotoxic waste, thereby creating a positive feedback loop that accelerates regional amyloid accumulation 2.
The Transition to Synaptic Failure and Atrophy
The early hyperactive phase imposes extreme metabolic demands on the cholinergic architecture. Chronic overstimulation induces severe metabolic stress, overwhelming the mitochondria and the endosomal-lysosomal pathways within the vulnerable basal forebrain neurons 2. Compounding this stress, alterations in the ubiquitin-proteasome system (UPS) driven by dysregulated nerve growth factor (NGF) and TrkA signaling pathways trigger an early "death" degradation process in cholinergic neurons prior to overt cellular apoptosis 26.
Concurrently, as Aβ levels transition from soluble oligomers to dense fibrillar plaques, their functional influence shifts from excitatory to inhibitory, aggressively suppressing cholinergic neurotransmission 2. Structural neuroimaging of patients across the AD continuum reveals progressive, quantifiable atrophy of the Ch4 subfields, with substantial volume reductions emerging as early as the subjective cognitive decline phase 78. Hyperphosphorylated tau directly infiltrates the nucleus basalis of Meynert, mechanically disrupting axonal transport and severing the critical neuromodulatory connections to the neocortex 24.
Direct Modulation of Amyloidogenesis by the M1 Receptor
The interaction between the cholinergic system and AD pathology is fundamentally bidirectional. M1 muscarinic receptors dictate the processing pathway of the amyloid precursor protein (APP) within cortical neurons. Activation of M1 shifts APP processing toward the non-amyloidogenic alpha-secretase pathway, which cleaves APP within the Aβ domain, preventing the formation of toxic Aβ peptides 2728. Conversely, when cholinergic input is lost and M1 receptors are left unactivated, the enzymatic balance shifts in favor of beta-site APP cleaving enzyme 1 (BACE1), accelerating the generation and aggregation of amyloid plaques 1827. Consequently, the progressive degradation of the BFCS effectively removes a critical biological brake on amyloidogenesis, accelerating the broader neurodegenerative cascade.
Genetic and Demographic Modulators of Cholinergic Decline
The manifestation of cholinergic decline and the clinical trajectory of AD display marked heterogeneity based on genetic polymorphism, demographic factors, and socio-economic status.
Choline Acetyltransferase Polymorphisms
Choline acetyltransferase (ChAT) is the primary enzyme responsible for synthesizing ACh from choline and acetyl-CoA. Genetic variants in the CHAT gene directly influence the basal rate of ACh synthesis, altering a patient's neurochemical baseline prior to the onset of neurodegeneration. Global epidemiological analyses indicate that specific CHAT polymorphisms modify AD risk, though penetrance exhibits significant regional and ethnic variation 2930.
The CHAT rs3810950 A/G missense mutation has been extensively studied. In Asian populations, the rs3810950 mutation shows significant negative associations with AD risk across multiple genetic models, suggesting specific allelic combinations alter disease susceptibility 293132. In Caucasian cohorts, the association is primarily observed under a recessive model or specifically in synergy with the APOE ε4 allele, where dual carriers present significantly accelerated cognitive decline 2930. Other polymorphisms, such as rs1880676 and rs2177369, have generally not been robustly validated as independent risk factors for AD across global meta-analyses 31.
Epidemiological Forecasts and Socioeconomic Risk Factors
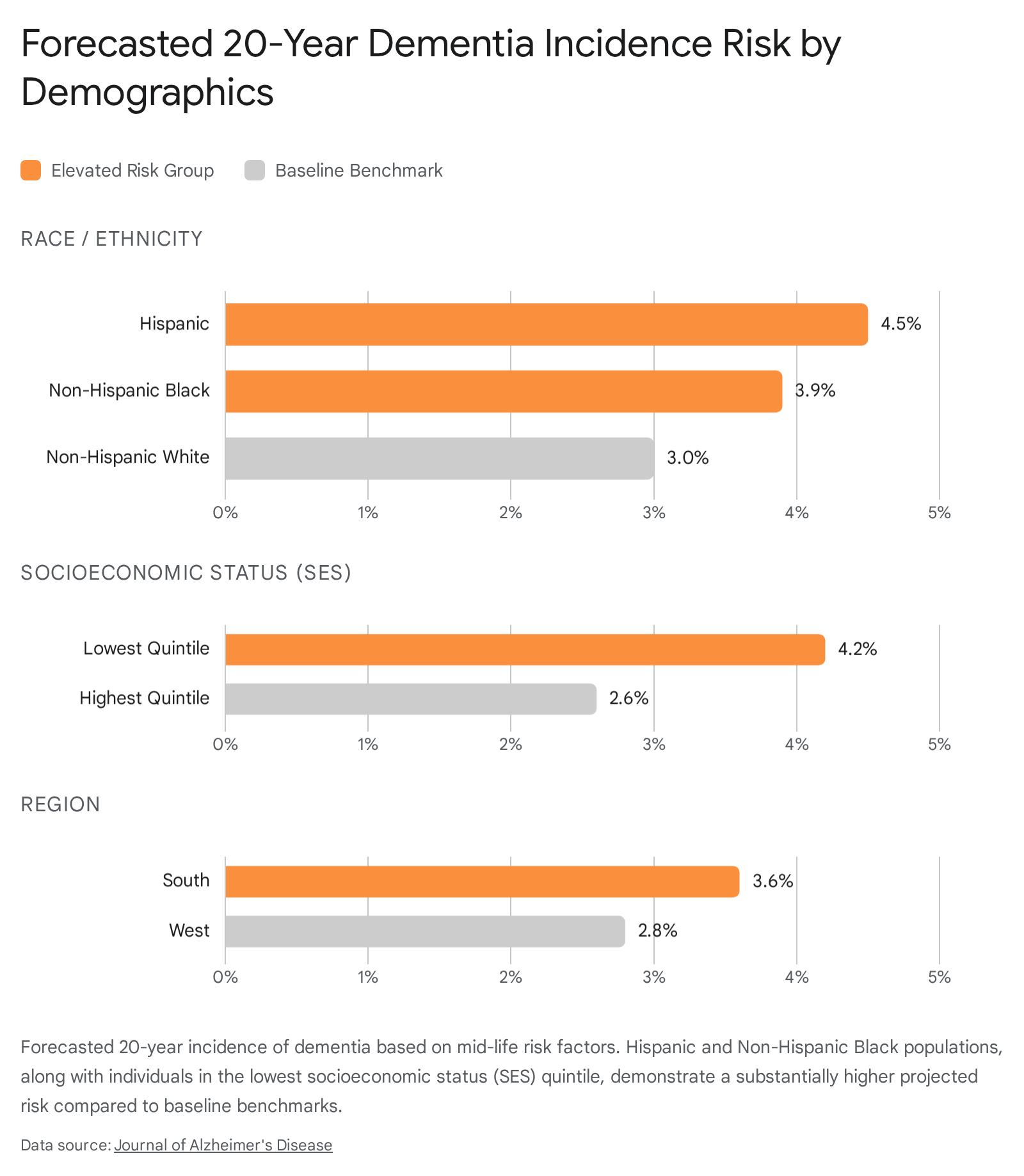
Epidemiological modeling demonstrates a stark disparity in the projected 20-year incidence of dementia across demographic groups. Based on mid-life risk profiles, Hispanic individuals exhibit an estimated incidence rate 1.51 times higher than Non-Hispanic White individuals, while Non-Hispanic Black individuals show a 1.27 times higher incidence 3334. Furthermore, socioeconomic status exerts a powerful independent effect; individuals in the lowest socioeconomic quintile face a 1.59 times higher risk of dementia incidence compared to those in the highest quintile 3335.
Biomarker Disparities and Trial Exclusions
These demographic differences extend into the underlying biology and biomarker presentation of the disease. In neuropathological and cerebrospinal fluid (CSF) analyses, African American patients with cognitive impairment frequently exhibit significantly lower baseline absolute levels of the tau protein compared to Caucasian patients with identical degrees of dementia severity 36.
Additionally, Black and Hispanic clinical trial participants are frequently excluded from advanced immunotherapies because they fail to meet the standardized amyloid threshold requirements, which were originally calibrated using predominantly White trial cohorts 34. These differences are hypothesized to stem partly from variations in the APOE ε4 gene, which acts as the single strongest genetic risk factor in White populations but exerts a markedly weaker amyloid-driving effect in populations of African and Hispanic ancestry 3436. As a result, standard diagnostic thresholds may inadvertently under-recognize AD pathology in minority populations, precluding them from potentially disease-modifying treatments.
Pharmacological Interventions and Clinical Disparities
For over two decades, the pharmacological management of cholinergic deficits relied exclusively on acetylcholinesterase inhibitors (AChEIs), including donepezil, rivastigmine, and galantamine 37. These agents operate by blocking the enzyme responsible for the synaptic degradation of endogenous ACh, thereby prolonging the residence time of the neurotransmitter in the synaptic cleft.
Limitations of Acetylcholinesterase Inhibitors
The efficacy of AChEIs is inherently rate-limited. Because they require the presence of endogenous ACh, their utility diminishes directly in parallel with the progressive degeneration of presynaptic cholinergic terminals in the basal forebrain 38. Moreover, AChEIs globally increase cholinergic signaling throughout the central and peripheral nervous systems, resulting in severe dose-limiting adverse effects, including gastrointestinal distress, bradycardia, and muscle cramping.
The utilization and persistence of AChEI therapies also reflect structural healthcare disparities. Retrospective analyses show that African American and Hispanic patients receive prescriptions for AChEIs at significantly lower rates than White patients, even after controlling for disease severity, age, and education 394041. Furthermore, among those who initiate AChEI therapy, medication non-adherence and discontinuation within the first year are profoundly higher in non-White cohorts 4142.
Pharmacogenomics of Cholinergic Therapy
The inter-individual variability in response to AChEIs is further compounded by pharmacogenomic factors. Cytochrome P450 enzymes, particularly CYP2D6 and CYP3A4, are responsible for the hepatic metabolism of donepezil and galantamine. Polymorphisms in these genes (such as the CYP3A41G non-carrier status) have been shown to correlate with poorer cognitive responses to AChEI therapy over a one-year period 43. Additionally, genetic variants in the butyrylcholinesterase (BCHE*) gene can influence the tolerability and efficacy of cholinesterase inhibition 3237. These pharmacogenomic variables, coupled with socioeconomic barriers to sustained care, result in minority populations bearing a disproportionate burden of unmitigated cholinergic decline.
Development of Next-Generation Cholinergic Therapeutics
To bypass the reliance on degenerating presynaptic terminals, modern drug development has shifted toward orthosteric agonists and positive allosteric modulators (PAMs) that directly target the postsynaptic muscarinic receptors (specifically M1 and M4). Because postsynaptic cortical receptors remain largely preserved even in advanced stages of the AD brain, these agents can theoretically maintain efficacy regardless of the functional integrity of the basal forebrain 272838.
| Investigational Asset | Mechanism of Action | Component Profile | Primary AD Clinical Targets | Trial Status (As of 2025/2026) |
|---|---|---|---|---|
| KarXT (Cobenfy) | M1 / M4 Orthosteric Agonist | Xanomeline + Trospium Chloride (Peripheral antagonist) | Psychosis (ADEPT), Cognition (MINDSET), Agitation (ADIAGO) | Phase 3 (ADEPT-2 results delayed to late 2026 due to site irregularities). |
| ML-007 (ML-007C-MA) | M1 / M4 Orthosteric Agonist | ML-007 + Fesoterodine (Extended-release peripheral antagonist) | Psychosis, Hallucinations, Delusions | Phase 2 (VISTA trial initiated mid-2025). |
| Emraclidine | M4 Positive Allosteric Modulator (PAM) | Single agent (Enhances endogenous ACh efficacy selectively) | Schizophrenia, Severe Agitation / Psychosis | Phase 3 (Demonstrated early efficacy in managing PANSS scores). |
Dual M1/M4 Orthosteric Agonists
The most advanced candidates in this class utilize a dual-component pharmacological strategy. The primary component is a highly penetrant, central muscarinic agonist that activates M1 and M4 receptors in the brain. The secondary component is a peripherally restricted anticholinergic agent (which cannot cross the blood-brain barrier) designed to selectively block the agonist's effects in the gut and cardiovascular system, thereby eliminating peripheral toxicity 2044.
KarXT (Cobenfy): KarXT is a fixed-dose combination of xanomeline, a potent M1/M4 orthosteric agonist, and trospium chloride, a peripheral muscarinic antagonist 4445. Originally validated for schizophrenia, KarXT achieved FDA approval in late 2024 as the first nondopaminergic antipsychotic, effectively managing positive and negative symptoms without utilizing dopamine D2 receptor antagonism 204446. Because an estimated 30% to 50% of AD patients suffer from delusions and hallucinations, KarXT is heavily investigated for Alzheimer's disease psychosis. It operates with a distinct advantage over atypical antipsychotics, as it avoids the black-box warnings for increased mortality, extrapyramidal symptoms, and cardiovascular events commonly associated with off-label antipsychotic use in elderly dementia patients 4446. Clinical evaluation is expanding under the ADEPT program; however, regulatory readouts for the critical ADEPT-2 phase 3 trial have been delayed to late 2026 following the discovery of site-execution irregularities that necessitated additional patient enrollment to restore statistical power 4647. Parallel Phase 3 global studies (MINDSET and ADIAGO) are exploring KarXT's efficacy in restoring broad cognitive function and mitigating severe agitation, respectively 4849.
ML-007 (ML-007C-MA): Addressing the rigid twice-daily dosing and fasting requirements of KarXT, MapLight Therapeutics developed ML-007C-MA as an extended-release formulation combining the novel M1/M4 agonist ML-007 with the peripheral antagonist fesoterodine 5052. ML-007 aims to offer a tightly synchronized pharmacokinetic profile that aligns central agonism with peripheral antagonism over an extended interval, allowing for convenient, food-independent dosing regimens 5051. Following successful Phase 1 trials demonstrating favorable safety and lack of significant cholinergic toxicity in elderly populations, the agent entered Phase 2 evaluation (the VISTA study) in 2025, directly targeting AD psychosis 5253.
Positive Allosteric Modulators
While dual M1/M4 orthosteric agonists address both cognitive decline (via M1) and behavioral symptoms (via M4), specific positive allosteric modulators (PAMs) are being developed to fine-tune cholinergic signaling with high subtype selectivity. Emraclidine, an M4-selective PAM, enhances the natural binding of endogenous ACh exclusively at the M4 receptor site 54. By acting allosterically rather than orthosterically, it preserves the temporal and spatial characteristics of physiological neurotransmission, avoiding the chronic receptor downregulation and desensitization often seen with direct agonists. Though primarily investigated and advanced to Phase 3 for schizophrenia, the mechanistic success of highly selective M4 modulation presents a viable future avenue for managing severe neuropsychiatric symptoms and agitation in AD without disrupting baseline cognitive functions governed by M1 receptors 2054.
Conclusion
The cholinergic system's regulation of attention and memory operates through a delicate, anatomically precise interplay between the basal forebrain and the neocortical strata. In Alzheimer disease, the breakdown of this system is not a linear late-stage failure but a complex, biphasic pathological arc. It begins with an initial, highly destructive period of amyloid-driven hyperactivity, progressing to the structural collapse of the nucleus basalis of Meynert and subsequent widespread neurotransmitter depletion. Because the loss of cortical M1 receptor activation directly promotes the generation of toxic amyloid via the BACE1 pathway, preserving cholinergic tone is fundamentally intertwined with modifying overall disease progression.
While legacy acetylcholinesterase inhibitors provided only limited symptomatic reprieve and highlighted deep demographic disparities in prescribing and adherence, the introduction of combination muscarinic agonists marks a paradigm shift in neuropharmacology. By surgically targeting preserved postsynaptic M1 and M4 receptors while actively blocking peripheral toxicity, these novel therapeutics offer the potential to safely restore cortical signal-to-noise processing, improve cognitive longevity, and mitigate the severe psychotic and agitated states that precipitate institutionalization. Future clinical and epidemiological advancements must ensure these pharmacological innovations are coupled with diagnostic frameworks calibrated for diverse demographic populations, ensuring that biological disparities in disease presentation do not limit access to emerging precision therapies.