What's Next for CRISPR Therapies in 2026
The 2026 CRISPR therapy landscape is defined by a rapid maturation from first-generation ex vivo treatments for rare blood disorders to advanced in vivo and epigenetic editing modalities targeting highly prevalent cardiovascular and autoimmune conditions. While clinical pipelines are accelerating and global regulators are actively modernizing approval pathways to accommodate these curative genetic medicines, widespread adoption remains severely bottlenecked by multimillion-dollar price tags, grueling patient treatment journeys, and insufficient healthcare infrastructure.
For the better part of a decade, the clustered regularly interspaced short palindromic repeats (CRISPR) system has been heralded in the public consciousness as a medical miracle waiting in the wings - a highly precise molecular tool destined to cure devastating, ultra-rare genetic diseases that have long eluded traditional pharmacology. By 2026, the scientific and commercial narrative has dramatically shifted. The underlying technology has advanced beyond ultra-rare orphan indications and is now systematically targeting some of the most common drivers of global morbidity and mortality, including atherosclerotic cardiovascular disease, resistant hypertension, and prevalent autoimmune disorders 123. However, as the clinical target population widens from the thousands to the millions, healthcare systems are colliding with an unforgiving economic reality. With initial treatments carrying list prices ranging from $373,000 up to an astronomical $4.25 million per patient, the transition of CRISPR from a niche scientific marvel to standard-of-care medicine hinges not merely on whether the underlying biology works, but on whether modern healthcare infrastructure can afford, reimburse, and distribute it equitably 345. The promise of a one-time cure is increasingly overshadowed by the realization that delivering these therapies requires a total rewiring of how medicine is manufactured, regulated, and financed.
How has CRISPR evolved beyond the original 'scissors'?
Since the initial demonstration of CRISPR-Cas9 for targeted genome editing, the biotechnology sector has experienced a rapid proliferation of new modalities, each designed to overcome the fundamental limitations of early-generation tools. The original CRISPR-Cas9 system is highly effective at disrupting genes but is inherently a blunt instrument. It can be likened to using scissors to edit a newspaper article: it is easy to cut out entire words or sentences, but difficult to surgically alter or replace individual letters without causing broader damage to the surrounding text 4.
When the traditional Cas9 enzyme identifies a target sequence using a guide RNA, it induces a clean double-strand break across the DNA double helix 56. The human cell responds by rushing to repair the catastrophic damage using an endogenous process known as non-homologous end joining (NHEJ). This repair pathway is notoriously inaccurate, frequently resulting in small, random insertions or deletions (indels) at the cut site 67. While this is highly useful for gene knockouts - intentionally disrupting a faulty gene so it no longer produces a disease-causing protein - it is highly suboptimal for precision corrections where a specific genetic sequence must be restored. To address this, researchers initially attempted to use homology-directed repair (HDR) by providing the cell with a "donor" DNA template, but the process of recruiting the appropriate donor DNA proved inefficient and prone to unwanted off-target effects, prompting the development of barcoded tracking platforms like MAGESTIC to improve cell survival and donor recruitment 48.
Ultimately, the drive for precision led the industry to advance toward next-generation modalities, which are best conceptualized through distinct, everyday analogies.
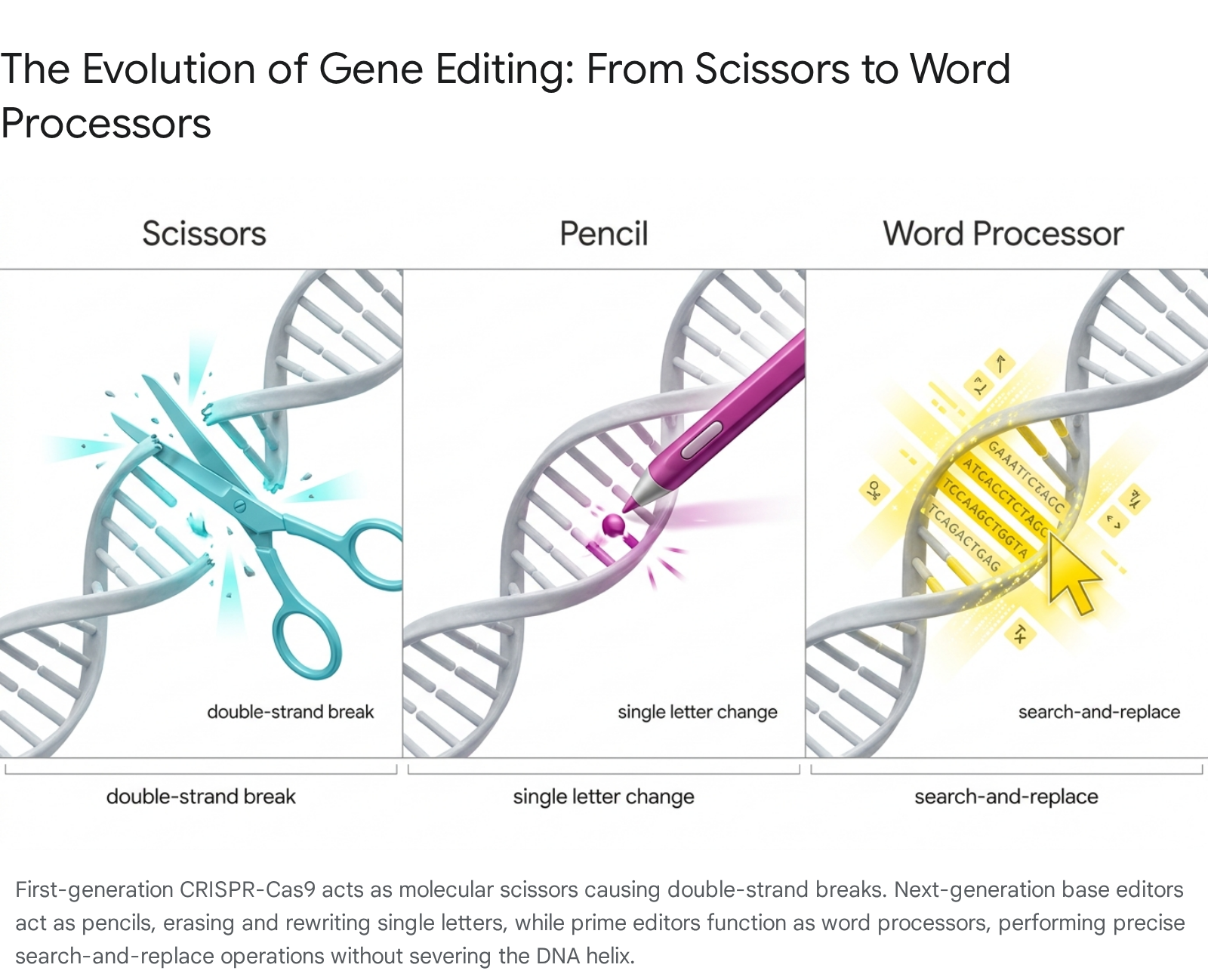
If Cas9 represents blunt scissors, base editors act as molecular pencils 5. These systems utilize a disabled, or "inert," Cas9 protein that can locate a precise target sequence but lacks the ability to sever the DNA strands. Instead, the disabled Cas9 is fused to a passenger enzyme - typically a deaminase - that chemically alters a single nucleotide base, such as converting an adenine (A) to a guanine (G), without breaking the double helix 611. This highly targeted approach is ideal for addressing diseases caused by single-point mutations, such as alpha-1 antitrypsin deficiency (AATD) or specific forms of familial hypercholesterolemia, offering pinpoint changes to understand or correct cellular function 369.
Advancing a step further, prime editing functions as a genetic word processor, enabling a highly efficient and exact "search and replace" capability 45. Utilizing a disabled SpCas9 protein fused with reverse transcriptase - an enzyme that transcribes RNA back into DNA - prime editors use a modified guide RNA (pegRNA) to both locate the target sequence and provide the exact replacement genetic code 10. Cryogenic electron microscopy has revealed how this unnatural combination of proteins maintains stability and achieves reverse transcription without cutting both strands of the double helix, allowing researchers to delete, insert, or swap longer stretches of genetic material with unprecedented accuracy and vastly reduced off-target liabilities 510.
Taking a fundamentally different approach, some next-generation developers are utilizing CRISPR systems that never permanently alter the underlying DNA sequence. This modality acts like a molecular dimmer switch. Companies such as Scribe Therapeutics are leveraging nuclease-inactivated CRISPR-CasX proteins fused to epigenetic effector domains. Rather than cutting DNA, these epigenetic silencers attach chemical marks to tune or turn off protein production from specific genes 13. For example, by targeting the PCSK9 gene to durably lower LDL cholesterol, this approach combines the precision and durability of CRISPR with the reversibility and perceived safety of RNA interference (RNAi), allowing the underlying genome to remain fully preserved 13.
Is CRISPR just a simple injection?
A prevailing and dangerous misconception in the public domain is that receiving a CRISPR therapy is akin to receiving a standard vaccine or an intravenous biologic drug. In reality, the patient experience, logistical infrastructure, and manufacturing requirements differ drastically depending on whether the therapeutic strategy utilizes an ex vivo or an in vivo approach.
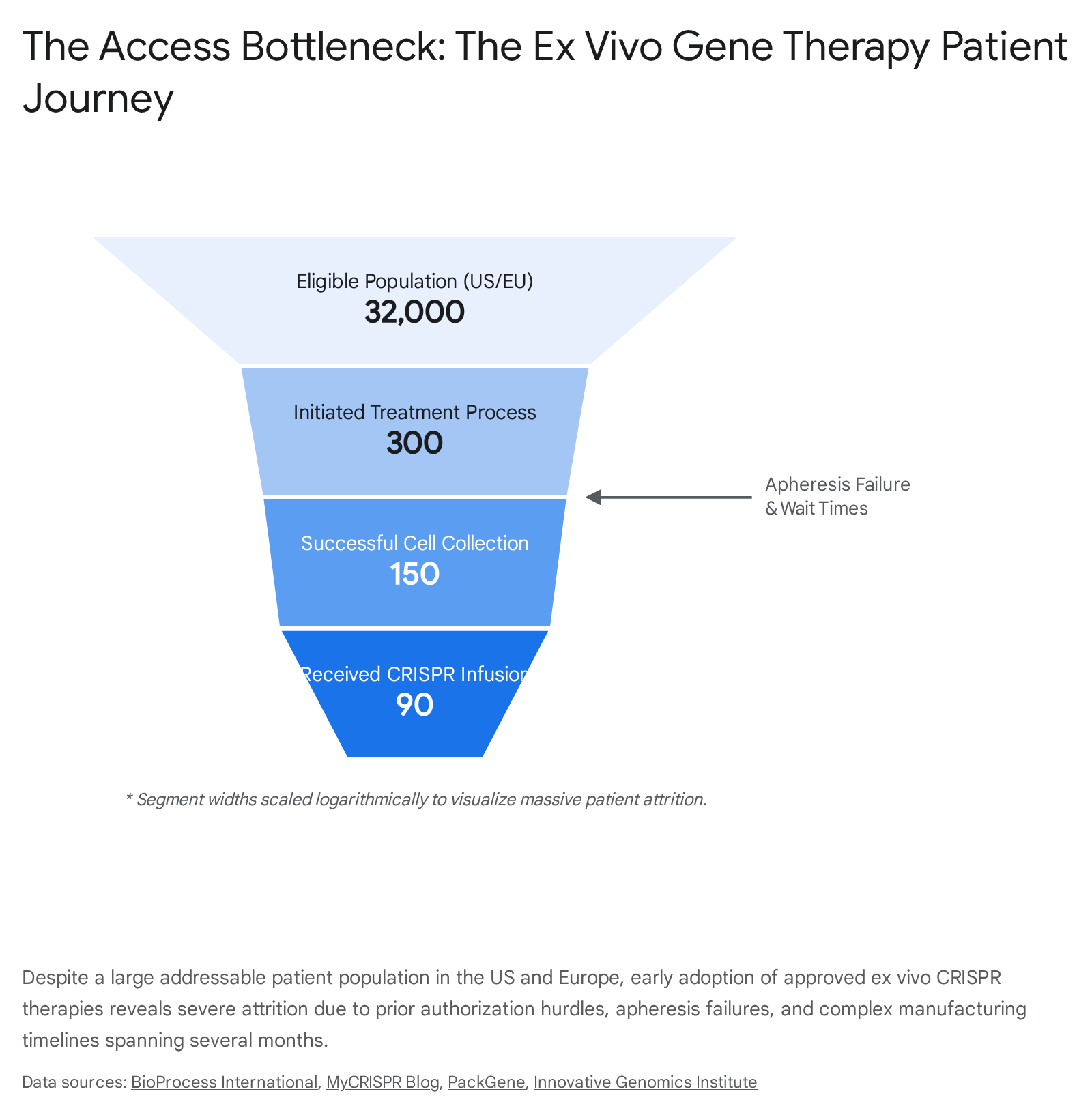
The currently approved vanguard of CRISPR therapeutics, notably Casgevy (exagamglogene autotemcel) developed by Vertex Pharmaceuticals and CRISPR Therapeutics for sickle cell disease (SCD) and transfusion-dependent beta-thalassemia (TDT), relies entirely on an ex vivo methodology 714. Ex vivo gene editing is not a simple injection; it is a grueling, highly choreographed cellular engineering marathon that can stretch over an entire year. The journey begins with the extraction of the patient's own hematopoietic stem cells through a process called apheresis 715. Because the subsequent laboratory processes - including electroporation to introduce the CRISPR machinery - can damage or kill a significant fraction of the harvested cells, clinical centers require a massive cellular yield to manufacture a viable dose 5. In practice, this first step has proven unexpectedly difficult; some patients with SCD have required up to five separate, exhausting hospital admissions spanning multiple weeks just to collect enough viable stem cells, leading a percentage of patients to abandon the process altogether 511.
Once sufficient cells are successfully collected, they are shipped to a centralized, specialized manufacturing facility. There, the CRISPR-Cas9 system is deployed to edit the BCL11A gene, modifying the stem cells to produce high levels of protective fetal hemoglobin 17. While the bespoke manufacturing takes several weeks, the most perilous phase of the patient journey remains. Before the edited cells can be reinfused, the patient must undergo myeloablative conditioning - intense, systemic chemotherapy using highly toxic agents like busulfan - to completely eradicate their existing, defective bone marrow 712. This conditioning phase carries severe, lifelong risks, including the high likelihood of permanent infertility, and requires the patient to remain hospitalized or isolated near the administering hospital for four to six weeks to manage potentially life-threatening side effects, severe pain, and profound immune vulnerability 1512. Only after the bone marrow is cleared are the gene-edited cells returned via infusion. While rival lentiviral vector therapies, such as bluebird bio's Lyfgenia, follow a similar ex vivo path, they carry additional boxed warnings from the FDA regarding the risk of blood cancer due to viral integration, highlighting the unique safety profiles patients must navigate 1112.
The long-term future of the field, however, rests on the realization of in vivo genome editing. In this approach, the instructions for the CRISPR gene editor are injected directly into the patient's body, eliminating the need to extract cells, manufacture them externally, or subject the patient to toxic myeloablative chemotherapy 7. By encapsulating the CRISPR machinery within lipid nanoparticles (LNPs) or utilizing adeno-associated viral vectors (AAVs), therapies can be administered via a single intravenous infusion 3131422. LNPs have a natural tendency to accumulate in the liver, making them highly effective for targeting hepatic genes, while AAVs are being aggressively developed to penetrate the central nervous system, retina, and muscular tissues 313. In 2026, in vivo approaches are reaching late-stage clinical maturity, offering a faster, more cost-effective, and dramatically less burdensome patient experience that more closely resembles traditional biologic administration 1415.
Table 1: Ex Vivo vs. In Vivo CRISPR Approaches 3571314221516
| Feature | Ex Vivo (First-Generation) | In Vivo (Next-Generation) |
|---|---|---|
| Mechanism of Action | Cells extracted, edited in a laboratory, and reinfused. | CRISPR machinery injected directly into the patient for in situ editing. |
| Delivery Vehicle | Electroporation utilized in a highly controlled cGMP lab. | Lipid Nanoparticles (LNPs) or Viral Vectors (AAVs). |
| Patient Burden | Immense. Requires apheresis, harsh chemotherapy (conditioning), and long hospital stays. | Low to Moderate. Standard IV infusion in an outpatient or clinical setting. |
| Timeline | 6 to 12 months from cell collection to final recovery. | Days to weeks (infusion plus short-term monitoring). |
| Manufacturing Model | High-variable-cost, "scale-out" bespoke manufacturing per patient. | High-fixed-cost, "scale-up" batch manufacturing. |
| Primary Target Tissues | Hematopoietic stem cells (blood) and immune cells (T-cells). | Liver (readily absorbs LNPs), retina, central nervous system, muscle. |
| Commercial Example | Casgevy (Sickle Cell Disease & Beta-Thalassemia). | Lonvo-z (Hereditary Angioedema - Anticipated 2027). |
What does the 2026 drug pipeline look like?
The 2026 clinical pipeline reflects a strategic and expansive pivot for the gene-editing industry. While rare monogenic disorders remain the foundational proving ground, developers are aggressively advancing both ex vivo and in vivo therapies for highly prevalent conditions, including hypercholesterolemia, systemic lupus erythematosus (SLE), and diverse oncological indications 317.
Intellia Therapeutics is currently spearheading the transition to commercial in vivo editing. Following the unprecedented success of its HAELO Phase 3 study for lonvoguran ziclumeran (lonvo-z) - designed to treat hereditary angioedema (HAE) by editing genes directly in the liver - the company initiated a rolling Biologics License Application (BLA) submission with the US FDA in 2026, targeting a commercial launch in the first half of 2027 1526. The HAELO trial data demonstrated an 87% reduction in HAE attacks following a single dose, with 62% of treated patients remaining entirely attack-free, proving that direct in vivo editing can achieve functional cures 15. Simultaneously, Intellia is advancing nex-z, targeting transthyretin amyloidosis, through a massive 1,200-patient global Phase 3 trial aimed at addressing a patient population estimated between 250,000 and 500,000 individuals globally 15.
CRISPR Therapeutics has leveraged its commercial momentum with Casgevy - which surpassed $100 million in revenue in 2025 and is currently seeking global regulatory label expansion into pediatric populations aged 5 to 11 - to fund a deep, diversified internal pipeline 227. The company expects critical mid-2026 data readouts for its allogeneic CAR-T candidate, zugocabtagene geleucel (zugo-cel), which is demonstrating early efficacy in both B-cell malignancies and autoimmune diseases like SLE, where early patients have maintained drug-free clinical remission with sustained B cell depletion 217. Furthermore, CRISPR Therapeutics is pushing in vivo candidates CTX310 and CTX320 into advanced trials for severe cardiovascular disease, alongside CTX460 for AATD and an emerging siRNA portfolio 215. Rival Beam Therapeutics is concurrently running pivotal trials for its base-editing platform (BEAM-101/risto-cel) and advancing its LNP-delivered in vivo base editor for AATD-1 3515. Other notable developments include Scribe Therapeutics initiating a Phase 1 study for STX-1150, an epigenetic silencing therapy engineered to durably reduce LDL-C without permanently altering the underlying DNA, aiming to redefine cardiovascular disease prevention 1.
A major defining dynamic of the 2026 pipeline is the explosive rise of the Chinese biotechnology sector, which is moving with unprecedented speed to capitalize on novel editing platforms and rapidly transitioning from preclinical discovery to advanced human trials. YolTech Therapeutics recently closed a $45 million Series B financing round to advance its base-editing and CRISPR-Cas therapies. The company is actively pushing YOLT-201, a CRISPR-based therapy for transthyretin amyloidosis, through Phase 1/2a trials, and notably secured IND clearance in both the US and China for YOLT-101, the first in vivo base-editing program for familial hypercholesterolemia 9. HuidaGene Therapeutics has bypassed standard Cas9 altogether, utilizing its AI-driven HG-PRECISE platform to deploy novel, proprietary editors. HuidaGene is advancing the DNA editor hfCas12Max for Duchenne muscular dystrophy (DMD) and the RNA-targeting hfCas13Y for MECP2 Duplication Syndrome, achieving early clinical milestones in investigator-initiated trials and utilizing short-lived RNA delivery vectors to minimize the risks of long-term Cas expression 2818. Similarly, Beijing-based EdiGene continues to advance its ATHENA CAR-T therapies for B-cell malignancies and SLE, utilizing SPPL3-null and T-cell receptor-deficient engineering to enhance safety and persistence, alongside its proprietary LEAPER RNA-editing platforms 3031.
Table 2: Snapshot of the 2026 CRISPR Clinical Pipeline 1239152630
| Drug Candidate | Company | Modality | Primary Indication | Current Clinical Status (2026) |
|---|---|---|---|---|
| Casgevy | Vertex / CRISPR Tx | Ex Vivo CRISPR-Cas9 | SCD, Beta-Thalassemia | Approved (Adults); Seeking Pediatric Expansion |
| Lonvo-z | Intellia Therapeutics | In Vivo CRISPR-Cas9 | Hereditary Angioedema (HAE) | Phase 3 Successful; BLA Submission Ongoing |
| Nex-z | Intellia Therapeutics | In Vivo CRISPR-Cas9 | Transthyretin Amyloidosis | Phase 3 Ongoing (1,200 patients) |
| STX-1150 | Scribe Therapeutics | In Vivo Epigenetic | ASCVD / High LDL-C | Phase 1 Ongoing |
| Zugo-cel | CRISPR Therapeutics | Ex Vivo Allogeneic CAR-T | Systemic Lupus (SLE), Oncology | Phase 1/2 Ongoing |
| BEAM-101 | Beam Therapeutics | Ex Vivo Base Editing | Sickle Cell Disease | Pivotal Trials Ongoing |
| YOLT-101 | YolTech Therapeutics | In Vivo Base Editing | Familial Hypercholesterolemia | IND Cleared (US & China) |
| HG-PRECISE (hfCas12Max/13Y) | HuidaGene Therapeutics | In Vivo DNA/RNA Editing | DMD, MECP2 Duplication | Phase 1/Investigator Trials Ongoing |
| ATHENA CAR-T | EdiGene | Ex Vivo Allogeneic CAR-T | B-NHL, SLE | Phase 1/2 Ongoing |
What are the contested unknowns of gene editing?
Despite transformative efficacy data and an accelerating pipeline, the deployment of gene therapies at a population scale is haunted by unresolved biological unknowns. The perception that CRISPR therapies ought to possess near-zero off-target effects belies the reality of clinical medicine; developers and regulators are increasingly recognizing that perfect therapeutics do not exist 19. Chief among these contested unknowns are the long-term durability of the edits and the profound complexities of off-target genotoxicity 2021.
The core value proposition of a multi-million-dollar CRISPR therapy is its permanence; a single administration is theoretically meant to provide a lifelong, durable cure 1113. However, the field currently lacks definitive longitudinal data extending beyond a few years of patient follow-up. A critical debate in 2026 revolves around the persistence of the delivery vectors and the resulting expression window of the Cas nucleases. Viral vectors, such as AAVs, can persist in human cells for years, offering sustained therapeutic expression but carrying elevated risks of long-term immune reactions, immunogenicity, or insertional oncogenesis 71321. Because CRISPR proteins originate from microbes, long-term expression might cause the human immune system to attack the edited cells, nullifying the therapy's benefits 18. Conversely, non-viral synthetic systems like LNPs degrade rapidly within hours or days 13. If a therapy relies on transient LNP delivery, will the edited stem cell populations eventually deplete or be outcompeted by unedited cells over a patient's lifespan? While current evidence in murine models, non-human primates, and early clinical cohorts suggests robust durability and manageable safety profiles, regulators globally acknowledge that declining functional transgene expression and the inability of gene addition alone to permanently modify disease-driving defects remain theoretical, long-term risks 1321.
The second major unknown is the full scope of off-target effects. While on-target editing effectively disrupts disease pathology, researchers are documenting a wide array of unintended genomic alterations resulting from CRISPR-Cas nuclease activity 722. These include small insertions and deletions (indels) as well as catastrophic structural variations (SVs) such as large deletions, inversions, and chromosomal translocations that could inadvertently silence tumor suppressor genes or trigger cellular death and secondary cancers 1922. Furthermore, recent peer-reviewed findings in 2026 highlight a newly identified phenomenon termed "chromatin fatigue." Studies indicate that even after a targeted DNA double-strand break is successfully repaired by the cell, the surrounding genome does not fully recover 36. Instead, the chromatin organization can remain structurally altered, leading to disrupted gene activity and heritable changes in gene expression that persist in future generations of cells 36. This lasting damage escapes standard safety assays focused solely on sequence fidelity, highlighting a serious layer of risk that is not currently accounted for in many assessments of gene editing technologies 36.
How are global regulators adapting to CRISPR therapies?
Regulatory agencies worldwide are scrambling to modernize legal frameworks originally designed for small molecules and traditional biologics, adapting them to the highly personalized, permanent, and complex nature of genetic engineering.
United States FDA: In April 2026, the US FDA released finalized draft guidance mandating rigorous new safety assessment standards for human gene therapy products that incorporate genome editing technologies. The agency now explicitly requires sponsors to utilize state-of-the-art next-generation sequencing (NGS) methodologies and advanced bioinformatics to track off-target editing risks and evaluate the loss of genome integrity in both ex vivo and in vivo platforms 3738. This science-based roadmap is intended to support Investigational New Drug (IND) and Biologics License Application (BLA) submissions 37. The FDA has firmly maintained a mandatory 15-year post-treatment follow-up period for all gene therapy recipients to monitor for delayed adverse events, such as secondary malignancies and autoimmune complications, mirroring prior guidance for traditional gene therapies 3940. However, to accelerate pipeline velocity, the FDA has simultaneously relaxed certain rigid Chemistry, Manufacturing, and Controls (CMC) specifications during Phase 2 and 3 trials, recognizing that excessive, inflexible manufacturing burdens stymie the development of individualized treatments 4041.
United Kingdom MHRA: The MHRA has emerged as one of the most progressive regulatory bodies for advanced therapies. In 2026, the agency launched a joint consultation to modernize the legal definition of Gene Therapy Medicinal Products (GTMPs). Recognizing that requiring GTMPs to be biological in origin excludes synthetic nucleic acid constructs that function identically, the MHRA is moving toward a mechanism-of-action definition that accommodates emerging non-recombinant platforms and base editors 4243. Furthermore, inspired by the success of personalized CRISPR therapies tailored to specific individuals - such as a customized treatment for a rare urea cycle disorder - the MHRA is establishing an "investigational marketing authorization" framework. This pathway grants a single overarching approval for platform technologies (e.g., a standardized lipid nanoparticle and Cas enzyme construct) containing a variable, tailored component, radically reducing the regulatory barrier for bespoke, N-of-1 medicines while maintaining strict real-world safety monitoring 4445. The MHRA also launched a joint pathway with the National Institute for Health and Care Excellence (NICE) on April 1, 2026, to synchronize licencing and value assessments, cutting market access times by up to six months 23.
Europe EMA: The EMA's Committee for Medicinal Products for Human Use (CHMP) continues to grant conditional authorizations for breakthrough gene therapies - including Casgevy - which mandate that manufacturers supply comprehensive post-market evidence to confirm long-term safety and efficacy 2425. Under the impending European pharmaceutical legislation reform, the Advanced Therapy Medicinal Products (ATMP) framework is undergoing significant revision. The EMA is heavily focused on refining hospital exemption rules, updating orphan drug designations to ensure they target true unmet medical needs rather than broad disease categories, and tightening long-term patient registry requirements to balance rapid access with stringent pharmacovigilance 40. The agency is also advancing the use of virtual control groups to reduce non-clinical animal testing 26.
China NMPA: The National Medical Products Administration (NMPA) has aggressively positioned China as a premier destination for gene therapy innovation, leveraging regulatory agility to foster a booming domestic ecosystem. Building on its 2017 ICH membership and 2020 reforms granting 30-day implied IND approvals, the NMPA enacted "Announcement No. 3" in 2026 505152. This policy moves away from gated, pre-approved lists for urgently needed overseas drugs to a more dynamic, applicant-initiated pathway that significantly accelerates clinical trial evaluations 50. The NMPA's Priority Review pathway targets a hyper-accelerated 130-working-day timeline (roughly 6 months) for novel NDAs, compared to 12 - 18 months for standard review, and the agency is increasingly utilizing Real-World Data (RWD) and conditional approvals 5152. Furthermore, a new law taking effect in May 2026 grants orphan drugs seven years of market exclusivity, incentivizing rapid development 53. This regulatory speed, combined with vast treatment-naive patient populations and a strong preference for multi-regional clinical trial (MRCT) integration, has allowed companies like HuidaGene and YolTech to bypass traditional delays and lead globally in early-stage in vivo trials 5154.
What this means for patients
The scientific validation of CRISPR therapies means little if the treatments never reach the patients whose lives depend on them. Currently, the gap between regulatory approval and real-world patient access is a formidable chasm defined by systemic infrastructure failure, astronomical costs, and rigid insurance paradigms that were never designed for single-dose curative medicines 5556.
The Infrastructure Bottleneck: For ex vivo therapies like Casgevy, the healthcare system is attempting to funnel tens of thousands of patients with systemic diseases into high-performance delivery architectures originally designed for a handful of rare blood cancers 57. Only a few hundred large academic medical centers globally have the accreditation, specialized staff, and resources to manage complex cellular handling, stem cell transplantation, and myeloablative conditioning 1257. While Vertex has successfully activated over 65 Authorized Treatment Centers (ATCs) globally, patients frequently face debilitating geographic barriers 56. The requirement to live within a two-hour radius of a transplant center for four to six weeks during conditioning effectively makes geography a form of triage, severely limiting access for rural or low-income populations and resulting in a plodding adoption rate during the first year of market availability 41257.
The Insurance and Cost Reality: At $2.2 million per course, ex vivo therapies pose an existential threat to traditional insurance risk pools 3527. While independent analyses, such as those conducted by the Institute for Clinical and Economic Review (ICER), suggest a subjective value of around $900,000 per treatment, market pricing remains intensely inflated due to exorbitant manufacturing costs and prolonged regulatory routes 28. For state Medicaid programs - which cover a disproportionate share of the SCD population - the Centers for Medicare & Medicaid Services (CMS) has launched the Cell & Gene Therapy Access Model 355. Under this model, state programs enter into outcomes-based agreements, paying drug manufacturers based on whether the therapy successfully improves health outcomes and reduces ongoing medical costs over time 360. Federal legislation, such as the Medicaid VBPs for Patients Act (MVP Act), is also being championed to modernize the framework for value-based purchasing arrangements, enhancing affordable access 27.
However, in the commercial and Medicare sectors, patients are exposed to crushing out-of-pocket costs due to standard deductibles and coinsurance designs, creating financial barriers for families already burdened by illness-related economic strain 429. Health economists estimate that the total annual cost of covering all expected cell and gene therapies could amount to less than $20 per member per month across the US population, but insurers are aggressively relying on reinsurance markets to protect themselves against unexpected, catastrophic claims 30. By 2026, the estimated reinsurance premium surcharge to cover gene therapies has reached nearly $15.69 per member per month 30. Payer strategies to mitigate this financial risk include highly restrictive prior authorization requirements, prolonged approval timelines, and out-of-network designations 55. Over 80% of healthcare providers report that persistent payer-related coverage issues delay care, with 60% noting that insurers enforce coverage criteria that exceed the FDA label language, preventing perfectly eligible patients from initiating treatment 55.
In the UK, access paradigms differ fundamentally. The National Institute for Health and Care Excellence (NICE) ultimately issued positive guidance for Casgevy, securing a reimbursement agreement with NHS England 17. This was facilitated through the Innovative Medicines Fund (IMF), a managed access scheme that allows the NHS to fund the £1.65 million therapy for up to 460 eligible patients immediately, while simultaneously collecting long-term real-world data to support future routine reimbursement decisions and tracking clinical and cost-effectiveness 141763.
The Hidden Barrier of Social Support: Strikingly, while clinical and financial barriers dominate the headlines, the day-to-day reality of gene therapy reveals a different, often overlooked primary obstacle. A comprehensive 2025 survey of over 100 advanced therapy providers identified that the lack of patient social and caregiver support (cited by 64% of respondents) and transportation challenges were the leading reasons patients failed to initiate therapy - outranking even insurance prior authorization denials (57%) and out-of-network designations (52%) 1555. Because the ex vivo process demands months of hospital visits, intense acute toxicity management, and prolonged recovery, patients without dedicated, full-time caregiver support are simply unable to navigate the logistical labyrinth and are often deemed clinically ineligible for treatment 15.
Bottom line
The 2026 CRISPR landscape represents a critical inflection point in the history of medicine. We have conclusively crossed the threshold from experimental theory to commercial reality, with foundational ex vivo platforms actively curing devastating genetic diseases and next-generation in vivo, base, and epigenetic editors poised to tackle the world's most prevalent chronic conditions 11516. Yet, this scientific triumph has exposed the profound inadequacies of the global healthcare delivery infrastructure.
The ultimate success of the CRISPR revolution will not be measured solely by the molecular precision of a lipid nanoparticle or the statistical significance of a Phase 3 trial 915. Instead, it will be judged by the industry's ability to transition from bespoke, high-friction ex vivo cellular manufacturing to scalable, off-the-shelf in vivo infusions, and the willingness of payers and regulators to forge novel financing models like the MVP Act and outcomes-based agreements 142760. Only by aggressively dismantling the geographic, financial, and logistical bottlenecks currently throttling patient access can the full curative promise of gene editing be equitably realized across the global population.