Telomere mechanisms in aging and the complexity of lengthening
Molecular Fundamentals of Telomere Architecture
Telomeres are highly conserved, specialized nucleoprotein structures situated at the termini of linear eukaryotic chromosomes. Their primary biological function is to protect internal genomic DNA from nucleolytic degradation, prevent the inadvertent fusion of adjacent chromosomal ends, and obscure the chromosomal termini from the cell's natural DNA double-strand break (DSB) repair machinery 12. Without these protective caps, the cellular DNA damage response (DDR) would erroneously identify natural chromosome ends as broken DNA strands, leading to cell cycle arrest, catastrophic chromosomal fusions, and widespread genomic instability 345.
In humans, telomeric DNA is composed of tandem, repetitive hexanucleotide sequences - specifically (TTAGGG)n. In normal somatic cells, these repeating sequences extend for approximately 5 to 15 kilobases (kb) 56. The structure terminates in a single-stranded 3' guanine-rich overhang that typically spans 50 to 500 nucleotides 57. This overhang folds back upon itself and invades the upstream double-stranded DNA region, forming a displacement loop (D-loop) within a larger, protective lariat-like structure known as a T-loop 5.
The physical stability and functional integrity of the telomere rely heavily on a specialized hexameric protein complex known as shelterin. The shelterin complex includes the double-stranded DNA-binding factors TRF1 and TRF2, the single-stranded DNA-binding protein POT1 (which sequesters the 3' overhang), alongside bridging and interacting factors such as TIN2, TPP1, and RAP1 5. By physically concealing the terminal DNA sequence, shelterin acts as a molecular cap that prevents the activation of DDR kinases, specifically ataxia-telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR), ensuring that chromosomal ends do not trigger anomalous senescence or apoptosis 589.
The End-Replication Problem and Telomere Attrition
The fundamental driver of age-related telomere attrition is the "end-replication problem." During the S-phase of the cell cycle, DNA polymerase synthesizes DNA strictly in a 5' to 3' direction. While the leading strand is synthesized continuously, the lagging strand is synthesized discontinuously via Okazaki fragments, which require RNA primers 71011. When the final RNA primer is removed from the extreme 5' end of the newly synthesized lagging strand, DNA polymerase cannot fill the resulting gap because it lacks a pre-existing 3'-hydroxyl group to extend 710.
Consequently, with every cycle of somatic cell division, human telomeres progressively shorten by an estimated 50 to 200 base pairs 1213. Over decades of human life, this steady, replication-driven attrition erodes the telomeric buffer. Once a telomere reaches a critically short length - often referred to as the Hayflick limit - the shelterin complex can no longer properly form or bind to the abbreviated sequence. The "uncapped" telomere is subsequently recognized as a critical double-strand break, triggering a sustained, p53- and Rb-mediated DNA damage response that forces the cell into an irreversible state of proliferative arrest known as replicative senescence 214.
High-Resolution Telomere Measurement Technologies
Historically, the study of telomere dynamics has been constrained by low-resolution measurement techniques that could only provide the average telomere length across all chromosomes in a cellular sample. However, recent technological advancements have revolutionized the granularity of telomere biology. A groundbreaking methodology known as Telo-seq, developed at the Salk Institute and published in 2024, permits researchers to determine the entire sequence and precise length of telomeres on each individual chromosome 1. Telo-seq has revealed that telomere shortening is incredibly heterogeneous; within a single human cell, each chromosome arm can exhibit vastly different telomere lengths and distinct shortening rates, likely influenced by chromosome arm-specific factors and localized mechanical stresses 1.
Other high-resolution techniques have also emerged. The Telomere length Combing Assay (TCA) allows for the precise measurement of telomere erosion on stretched DNA fibers without the need for cycling cells, demonstrating extreme sensitivity to telomere length changes of 1 kb or less 6. For malignancies, the Single Molecule Telomere Assay Optical Mapping (SMTA-OM) is now utilized to quantify individual telomeres and identify unique structural features - such as telomere-free ends and superlong telomeres - at the single-molecule level 15. Concurrently, computational frameworks like EXTEND have been validated for quantifying telomerase enzymatic activity directly from bulk RNA sequencing data, allowing researchers to stratify clinical samples into specific telomerase activity phenotypes 16.
Telomere Attrition Within the Hallmarks of Aging
Telomere attrition is universally recognized as one of the primary drivers of biological aging. In the expanded "12 Hallmarks of Aging" framework proposed by López-Otín and colleagues, updated in 2023, telomere attrition is classified as a "primary hallmark" - an inherently negative, initiating trigger that accumulates cellular damage progressively over time 171819. The 12 hallmarks include genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, disabled macroautophagy, deregulated nutrient-sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, chronic inflammation, and dysbiosis 18192021.
Primary hallmarks provoke downstream compensatory responses that eventually lead to organismal decline 1821. When telomere shortening forces a cell into replicative senescence, the cell does not immediately undergo apoptosis. Instead, senescent cells undergo profound metabolic and transcriptomic shifts, adopting the senescence-associated secretory phenotype (SASP) 82122. Cells exhibiting SASP excrete a localized barrage of pro-inflammatory cytokines (such as IL-6 and TNF-α), chemokines, and matrix metalloproteinases into the surrounding tissue microenvironment 4.
The accumulation of senescent cells over a human lifespan disrupts local tissue architecture, impairs stem cell niches, and promotes a state of chronic, low-grade systemic inflammation often termed "inflammaging" 21. This microenvironmental toxicity directly fuels secondary aging hallmarks, including stem cell exhaustion and altered intercellular communication, driving age-related pathologies ranging from neurodegeneration to cardiovascular disease, osteoporosis, and metabolic dysfunction 182123. Research has shown a direct correlation between the accumulation of telomere-associated DNA damage response foci and the disruption of autophagy, impaired proteostasis, and profound epigenetic dysregulation 23.
The Cause Versus Symptom Debate in Biological Aging
While leukocyte telomere length (LTL) is firmly established as a highly accurate biomarker of biological age, its exact role as a direct causative agent of all systemic age-related diseases is the subject of intense, ongoing scientific debate 132425. Traditional biological models posited that telomere attrition directly and universally drove tissue failure. However, high-resolution epidemiological studies suggest a more nuanced, bidirectional relationship.
Analyses of massive datasets, such as the UK Biobank and the All of Us Research Program (involving cohorts ranging from 356,000 to over 679,000 individuals), indicate that while short LTL is highly correlated with conditions like stroke, late-life depression, and dementia, it may not be the direct physiological cause of these neurological conditions 26272829. Instead, researchers increasingly suggest that LTL acts as a highly sensitive, reflective "dosimeter" of cumulative physiological damage, chronic inflammation, and oxidative stress over an individual's lifetime 2629.
This nuance is further reinforced by 2025 investigations into progeroid (premature aging) syndromes. A comparative study assessing DNA methylation and telomere length across multiple rare genetic aging disorders - including Werner Syndrome, Hutchinson-Gilford Progeria Syndrome, and Dyskeratosis Congenita - found that telomere attrition is not a universal characteristic of all progeroid phenotypes 30. While syndromes driven by specific DNA repair defects or telomerase mutations exhibit profound telomere damage, other conditions presenting with identical premature aging phenotypes do not demonstrate abnormal telomere shortening 30. This divergence explicitly demonstrates that while telomere shortening is a major pathway of aging, biological aging can proceed through parallel, telomere-independent molecular mechanisms.
Population Genomics and Telomere Heterogeneity
Telomere length is a highly heterogeneous trait, influenced by an intricate interplay of genetic inheritance, environmental exposures, and lifestyle factors. Mean telomere length demonstrates substantial heritability, with estimates ranging from 44% to over 84% depending on the specific cohort and cellular subset 123132.
Ancestral and Geographical Variations
Global genomic analyses, particularly the 2024 and 2025 multi-ancestry meta-analyses stemming from the All of Us Research Program, have revealed highly significant variations in LTL across different ancestral populations. Analyzing whole-genome sequences from over 242,000 diverse participants, researchers observed that individuals of African ancestry in the Americas (such as African Caribbean in Barbados and African Ancestry in Southwest USA cohorts) consistently display significantly different, and generally longer, constitutive telomere lengths compared to Eurasian and continental African populations 2733.
Furthermore, geographical heterogeneity is highly pronounced. Accounting for age, sex, and genetic ancestry, analyses of the United States population revealed that significantly longer leukocyte telomeres cluster geographically in the West Coast and Central Midwest, while significantly shorter telomeres cluster in the Southeast 28.
Genome-wide association studies (GWAS) and rare variant aggregation studies (RVAS) combining All of Us data with the UK Biobank have identified 234 non-overlapping genetic loci linked to human telomere length variations, including 37 entirely novel loci 272834. Many of these loci reside near established telomere biology genes such as TERT, TERC, OBFC1, and RTEL1 7273135. These genetic baseline differences mandate that two individuals of the exact same chronological age can possess vastly different biological reserves of telomeric DNA, establishing entirely divergent trajectories for cellular senescence based purely on inherited genomics.
Environmental and Biochemical Modulators of Telomere Integrity
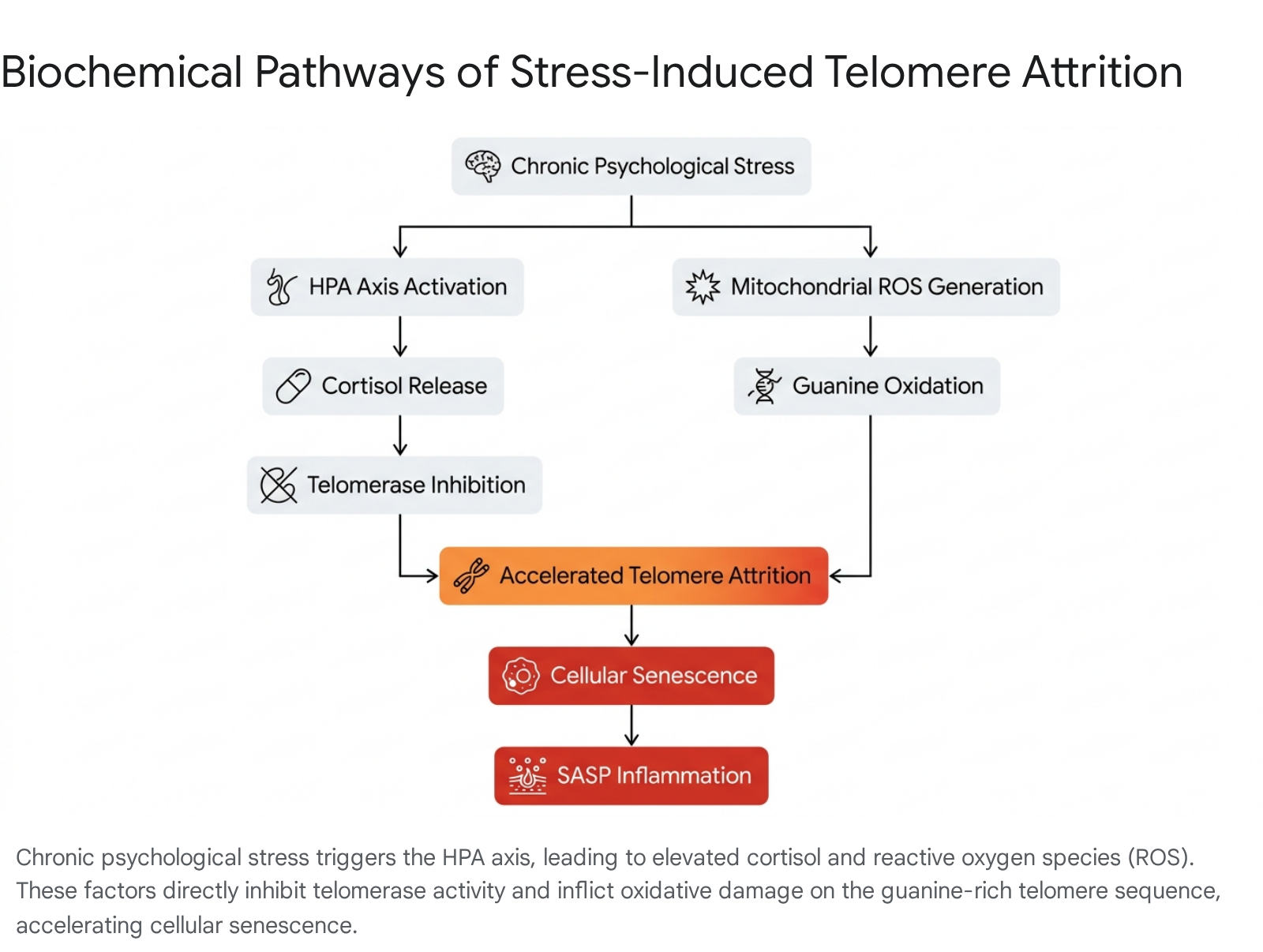
Beyond inherited genetics, the rate of telomere attrition is exquisitely sensitive to environmental and biochemical stress. Chronic psychological and physiological stress accelerates cellular aging through specific neuroendocrine pathways, primarily via the prolonged activation of the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic nervous system 436.
Glucocorticoid Exposure and Telomerase Inhibition
Sustained psychological stress results in continuously elevated systemic cortisol levels. Both in vitro and human longitudinal studies have demonstrated that chronic exposure to cortisol directly downregulates the activity of telomerase in peripheral blood mononuclear cells (PBMCs) and T lymphocytes 4836. Specifically, elevated cortisol reduces the transcription of hTERT, the catalytic component of telomerase, effectively crippling the cell's ability to maintain telomeric repeats under stress 48.
Oxidative Stress and DNA Damage Amplification
Simultaneously, chronic stress upregulates general metabolic activity and mitochondrial oxygen consumption, generating excessive reactive oxygen species (ROS) as a byproduct of accelerated oxidative phosphorylation 837. The specific guanine-rich sequence of telomeric DNA (TTAGGG) makes it exceptionally vulnerable to oxidative damage 14223238.
ROS induce single-strand breaks and guanine oxidation directly within the telomere sequence, disrupting the subsequent binding of the shelterin complex 2238. Because telomeric DNA is inherently difficult to replicate and repair compared to the rest of the genome, this damage is often persistent. Consequently, oxidative stress acts as a potent biochemical catalyst, accelerating telomere shortening and structural uncapping far beyond the baseline rate expected from the end-replication problem alone 41438.
Lifestyle Interventions and Telomere Preservation
Conversely, rigorous lifestyle interventions have been empirically linked to the preservation of telomere length. Cross-sectional data from the National Health and Nutrition Examination Survey (NHANES) evaluating 4,814 adults revealed that physical activity, specifically regular strength training, acts as a protective buffer against cellular aging 39. The analysis demonstrated that precisely 90 minutes of strength training per week was associated with significantly longer leukocyte telomeres, equating to an average reduction of 3.9 years in biological aging when adjusting for covariates such as socioeconomic status, body mass index, and diet 39. Optimal lifestyle parameters modulate the biological clock by dampening the systemic release of pro-inflammatory cytokines, optimizing mitochondrial efficiency, and reducing the total cellular load of reactive oxygen species 42337.
Telomere Maintenance Mechanisms in Oncogenesis
To bypass the limits of the end-replication problem, specific cellular lineages possess mechanisms to maintain or elongate their telomeres. The primary mechanism is the ribonucleoprotein enzyme complex known as telomerase. Telomerase consists of two main components: Telomerase Reverse Transcriptase (TERT), the catalytic protein subunit, and Telomerase RNA Component (TERC), a long non-coding RNA that serves as the internal template for the addition of de novo TTAGGG repeats 134041.
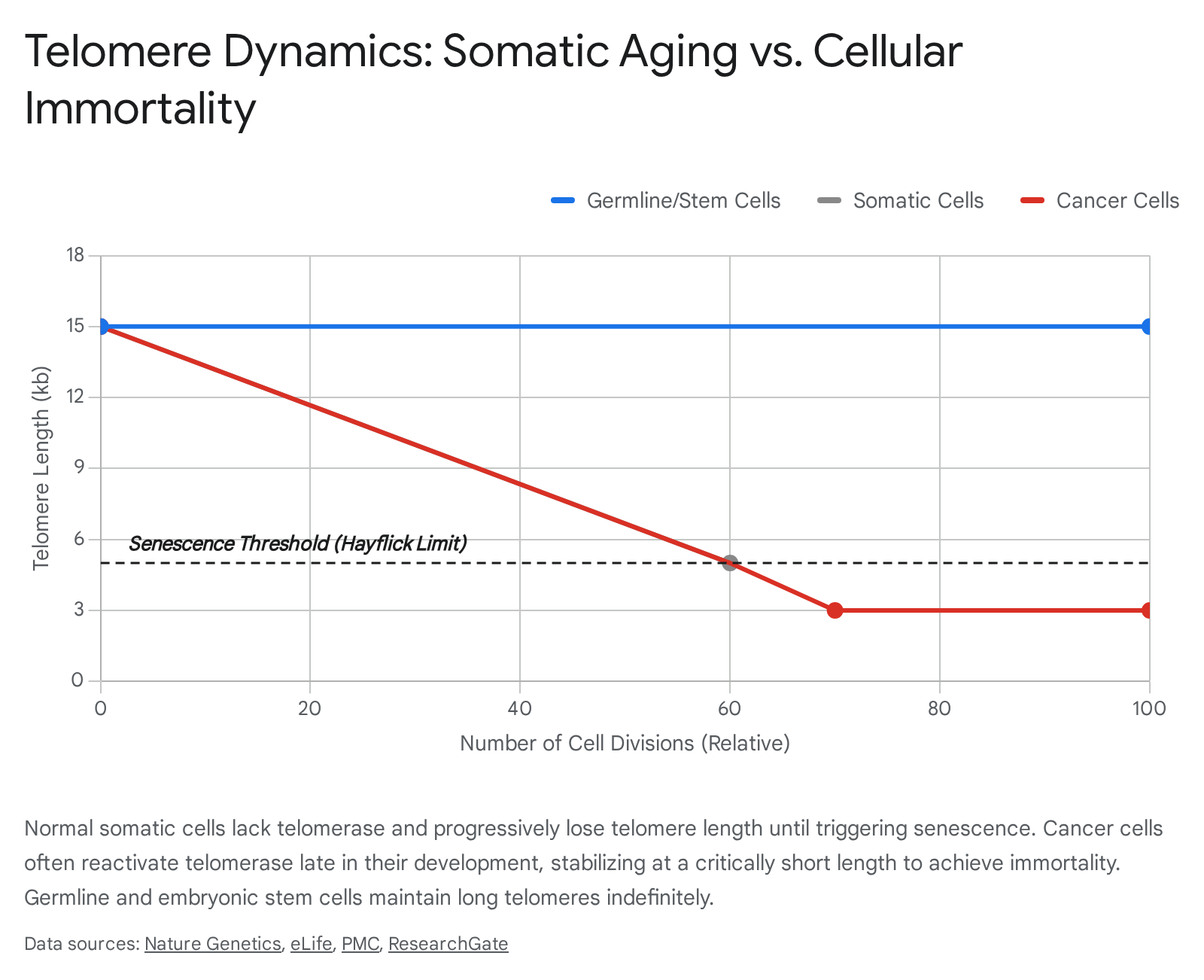
In humans, telomerase activity is highly context-dependent and strictly regulated based on cellular differentiation status. In the germline and in human embryonic stem cells (hESCs), telomerase is expressed at exceptionally high levels 134243. This continuous maintenance allows these cells to divide infinitely without telomere erosion, ensuring the passage of full-length genomes to subsequent generations 4445. In adult stem cells (such as hematopoietic stem cells), telomerase operates at a low, basal level that slows, but does not entirely prevent, telomere shortening 4446. However, in differentiated somatic cells, the TERT gene is heavily methylated and transcriptionally silenced. Somatic cells completely lack telomerase activity, rendering them subject to a strict biological clock 64044.
Telomere Dynamics by Cell Lineage
| Cell Classification | Baseline Telomere Length | Telomerase Expression | Replicative Capacity | Primary Biological Implication |
|---|---|---|---|---|
| Germline / Embryonic Stem Cells | 15 - 20 kb | Very High | Unlimited (Immortal) | Generational genetic transfer; embryogenesis 134345. |
| Adult Stem / Progenitor Cells | 8 - 12 kb | Low / Transient | High, but finite | Tissue renewal; subject to eventual exhaustion 4446. |
| Differentiated Somatic Cells | 5 - 10 kb (declining) | Undetectable / Silenced | Strictly limited (~50-70 divisions) | Specialized tissue architecture; susceptible to senescence 640. |
| Telomerase-Positive Cancer Cells | Highly variable (often short) | High (Reactivated) | Unlimited (Immortal) | Malignant proliferation and tumor progression 414748. |
Telomerase Reactivation in Cancer
The strict suppression of telomerase in somatic cells serves as an evolved, potent anti-cancer mechanism. By capping the number of times a cell can divide, the body creates a natural barrier against unchecked proliferation 240. To achieve malignancy, a cancer cell must successfully bypass replicative senescence by acquiring a Telomere Maintenance Mechanism (TMM) 340.
In approximately 85% to 90% of all human cancers, malignant cells achieve this by genetically or epigenetically reactivating the TERT gene 54041. This reactivation is frequently driven by TERT promoter mutations (highly prevalent in melanoma and glioblastoma), genomic rearrangements, DNA copy number amplifications, or altered DNA methylation 414749.
Paradoxically, despite extremely high telomerase enzymatic activity, the absolute telomere length in most solid tumors is often significantly shorter than in adjacent healthy tissue 24150. This phenomenon occurs because telomerase reactivation typically happens late in oncogenesis, after pre-malignant cells have undergone extensive, rapid division and their telomeres have already become critically short 4142. The reactivated telomerase simply maintains the telomeres at this shortened, fragile state - preventing mitotic catastrophe while permitting the ongoing genomic instability necessary for rapid cancer evolution 2942.
The Alternative Lengthening of Telomeres (ALT) Pathway
While the vast majority of tumors rely on telomerase, approximately 10% to 15% of human cancers achieve replicative immortality through a telomerase-independent mechanism known as the Alternative Lengthening of Telomeres (ALT) pathway 51051. The ALT pathway is uniquely prevalent in specific malignancies, notably soft-tissue sarcomas, isocitrate dehydrogenase (IDH)-mutant lower-grade gliomas, pancreatic neuroendocrine tumors, and pediatric neuroblastomas 31011.
The ALT mechanism operates by co-opting the cell's homologous recombination (HR) DNA repair machinery 31011. When an ALT-positive cell experiences a critically short or damaged telomere, it utilizes a process analogous to break-induced replication (BIR) known as break-induced telomere synthesis (BITS) 1151. The single-stranded 3' overhang of the damaged telomere invades the telomeric sequence of a neighboring sister chromatid or an extrachromosomal telomeric DNA circle (C-circle) 1151. Guided by proteins such as RAD52 and SLX4, DNA polymerase extends the broken telomere using the homologous sequence as a template 511.
This interchromosomal recombination is highly aberrant. As a result, ALT-positive cancers exhibit distinct clinical hallmarks: extreme cell-to-cell telomere length heterogeneity (with some telomeres measuring over 50 kb and others nearly absent), widespread chromosomal instability, and the presence of unique molecular markers like C-circles and ALT-associated promyelocytic leukemia bodies (APBs) 5101115. Genetically, ALT-positive tumors frequently harbor inactivating loss-of-function mutations in chromatin remodeling genes such as ATRX, DAXX, and H3F3A, which collectively permit this unregulated telomeric recombination 21011.
Therapeutic Interventions and Clinical Complexities
Given that telomere shortening tracks intricately with human aging and the exhaustion of tissue stem cells, reversing this process by therapeutically elongating telomeres has been a primary objective of regenerative medicine. However, manipulating telomere length in vivo is vastly more complex and fraught with biological risk than simply upregulating a deficient enzyme.
The Oncogenic Paradox of Telomere Elongation
The paramount barrier to systemic anti-aging telomerase therapies is the profound risk of carcinogenesis 2347. Somatic silencing of telomerase evolved specifically to protect long-lived mammals from cancer. Systemically delivering gene therapies that permanently reactivate the TERT gene poses an existential risk: it threatens to supply clinically silent, pre-malignant cells - which naturally harbor accumulated genetic mutations over an individual's life - with the final tool they need to evade senescence and achieve unbridled replicative immortality 2347.
Consequently, modern therapeutic design must navigate an exceptionally narrow biological window. Interventions must provide enough telomere extension to rejuvenate tissue stem cells without breaking the senescence barrier that suppresses occult tumors 50. Furthermore, structural revelations confirming that individual chromosome arms possess varying telomere lengths dictate that blunt-force, systemic telomerase activation could interact unpredictably with localized DNA damage responses 1.
Transient RNA Engineering and eTERC
To circumvent the risks associated with permanent genetic integration and continuous telomerase expression, researchers have recently pivoted to transient, "hit-and-run" molecular approaches.
In a landmark 2025 study, bioengineers at Boston Children's Hospital developed a highly innovative therapy utilizing engineered telomerase RNA (eTERC) 52535456. Delivering therapeutic long non-coding RNA (lncRNA) molecules has historically been challenging due to rapid intracellular degradation. However, scientists engineered a stabilized, synthetic lncRNA capable of substituting for the missing TERC component in patient stem cells 5455. Utilizing the non-canonical polymerase TENT4B to catalyze self-limited 2'-O-methyladenosine tailing, the researchers effectively shielded the 3' end of the synthetic RNA from rapid nucleolytic decay 5456.
Crucially, when this eTERC is introduced into human hematopoietic stem cells via electroporation, it provides a massive, temporary burst of telomerase activity. Within three days, target telomeres are extended by over 1 kilobase 56. Because the synthetic RNA eventually degrades naturally, the telomerase activity ceases, and standard cellular attrition mechanisms resume within approximately 69 days 5256. This brief intervention allows for a dramatic extension of the cell's replicative lifespan - extending it by up to 65 population doublings - without leaving a permanent genetic alteration that could fuel oncogenesis 545655.
Advancements in In Vivo Gene Therapy
Simultaneously, clinical trials targeting catastrophic telomeropathies have reached critical milestones. Telomere Biology Disorders (TBDs), such as dyskeratosis congenita, are progeroid-like diseases caused by inherited mutations in telomere-maintenance genes. Patients are born with critically short telomeres, leading to rapid bone marrow failure, pulmonary fibrosis, and early mortality 6315656.
In early 2025, the first in-human gene therapy trial for TBDs, overseen by Cincinnati Children's Hospital, reported highly promising phase 1/2 results 575861. The trial utilized an investigational therapy, EXG-34217, which employs the ZSCAN4 gene to drive in vivo telomere elongation specifically within hematopoietic stem cells 61. Data published in NEJM Evidence revealed that patients treated with the therapy achieved sustained in vivo telomere elongation in subpopulations of lymphocytes and granulocytes, successfully returning telomere lengths to the normal age-adjusted range 565861. Most notably, over follow-up periods extending up to 24 months, patients showed marked improvements in absolute neutrophil counts (ANC) without requiring the harsh preconditioning regimens (like chemotherapy or radiation) typical of stem cell transplants 5861. To date, no treatment-related safety concerns or oncogenic events have been observed, validating the potential of targeted, disease-modifying genetic interventions 565861.
Parallel experimental research involving high-capacity cytomegalovirus (CMV) vectors has also shown promise in murine models. By delivering exogenous TERT and follistatin via intranasal or intraperitoneal routes, researchers successfully extended median lifespans by up to 41%, halted mitochondrial structure deterioration, and improved glucose tolerance without an observable increase in cancer incidence 59. While human translation of these broader anti-aging viral therapies remains distant, they underscore the profound physiological leverage held within the telomere maintenance network.
Conclusion
Telomere attrition represents a profound biological intersection between the physical limits of DNA replication and the broader architecture of human aging. As a primary, initiating hallmark of aging, the progressive erosion of the chromosomal buffer enforces replicative senescence and drives the downstream SASP-mediated tissue degradation characteristic of systemic physical decline.
However, telomere biology is defined by a delicate, high-stakes evolutionary compromise. The cellular silencing of telomerase that guarantees our eventual physiological decay is the exact same mechanism that protects large, long-lived mammals from rampant malignancy during their reproductive years. The complexity of therapeutic intervention lies in navigating this paradox. Telomeres do not shorten uniformly, their attrition is heavily accelerated by environmental oxidative stress and HPA-axis dysregulation, and any blunt manipulation of their length is inherently tied to the oncogenic pathways hijacked by both telomerase-positive and ALT-positive cancers.
Recent clinical and bioengineering triumphs - ranging from transient RNA delivery systems like eTERC to targeted in vivo gene therapies like EXG-34217 - suggest that the scientific community is finally learning to thread this molecular needle. By shifting the paradigm away from permanent, systemic telomerase reactivation and toward highly controlled, localized elongations in targeted stem cell populations, researchers are unlocking the potential for safe, disease-modifying therapies that can mitigate the decay of human tissues without sparking the very cancers the telomere system evolved to prevent.