Systemic inflammation in Alzheimer's and Parkinson's diseases
Introduction
Historically, neurodegenerative disorders such as Alzheimer's disease and Parkinson's disease were viewed almost exclusively through a brain-centric lens, characterized primarily by localized neuronal loss and central protein aggregation. However, the accumulation of epidemiological, molecular, and clinical evidence has catalyzed a paradigm shift, reframing these conditions as systemic crises of proteostasis and immunity 1. At the core of this modern understanding is the concept of "inflammaging" - a chronic, low-grade, and sterile systemic inflammation that universally accompanies advancing age 23.
While inflammaging acts as a general physiological byproduct of cellular aging and environmental exposures, it is now recognized as a primary driver of neurodegeneration. This systemic inflammatory burden primes the innate immune system and disrupts the central nervous system, transforming homeostatic glial cells into agents of synaptic destruction and neuronal toxicity 24. The inflammaging-neurodegeneration axis describes the bidirectional and pathogenic communication between peripheral systemic inflammation and the neuroimmune infrastructure. Through compromised blood-brain barrier integrity and neural signaling routes, peripheral cytokines and immune cells breach the central nervous system, driving the onset, progression, and exacerbation of Alzheimer's and Parkinson's pathologies long before cognitive or motor symptoms become clinically apparent 15. This report evaluates the biological cascades, epidemiological modifiers, and emerging therapeutic interventions that define the inflammaging-neurodegeneration axis.
Drivers of Systemic Inflammaging
The onset of chronic systemic inflammation is multifactorial, arising from the cumulative effects of cellular exhaustion, lifelong pathogen exposure, metabolic dysfunction, and profound shifts in the host microbiome. These peripheral factors generate a sustained inflammatory tone that exerts relentless pressure on central nervous system resilience.
Peripheral Cellular Senescence
Cellular senescence is a state of irreversible cell cycle arrest triggered by varied stressors, including telomere attrition, genomic instability, epigenetic alterations, and oxidative stress 46. While senescent cells cease to divide, they remain highly metabolically active and adopt a senescence-associated secretory phenotype 78. This phenotype is characterized by the continuous, low-level secretion of pro-inflammatory cytokines, chemokines, growth factors, and matrix metalloproteinases 910.
In the periphery, the accumulation of senescent epithelial, endothelial, and immune cells creates a persistent inflammatory tone in the bloodstream. The chronic release of these secretory factors taxes the immune system, leading to immunosenescence - a gradual deterioration of the immune system's ability to mount targeted responses and resolve inflammation 411. Furthermore, cellular senescence exhibits a paracrine effect; senescent cells can induce senescence in neighboring healthy cells, accelerating tissue dysfunction and amplifying the systemic inflammatory signal that eventually reaches the brain 8. The peripheral accumulation of these cells ensures that circulating levels of potent cytokines, such as interleukin-6 and tumor necrosis factor-alpha, remain constitutively elevated in older adults, providing a continuous noxious stimulus to the neurovascular unit 59.
Gut Dysbiosis and the Microbiome
The human gastrointestinal tract harbors a complex microbial ecosystem that plays a foundational role in educating and regulating the systemic immune system. With age, the composition and diversity of the gut microbiome undergo significant alterations - a state known as dysbiosis 6. Dysbiosis is characterized by a reduction in beneficial, anti-inflammatory bacteria and an overgrowth of pathogenic or pro-inflammatory strains 12.
Beneficial microbes typically produce short-chain fatty acids, such as butyrate, propionate, and acetate, which are critical for maintaining the integrity of the intestinal epithelial barrier, modulating mitochondrial health, and exerting broad anti-inflammatory effects 7. A decline in short-chain fatty acid-producing bacteria compromises the intestinal mucosa, leading to increased gut permeability 9.
This breakdown of the intestinal barrier permits the translocation of microbial endotoxins - most notably lipopolysaccharides and bacterial amyloids, such as curli fibers - into the systemic circulation 59. Circulating lipopolysaccharides directly stimulate Toll-like receptor 4 on peripheral immune cells and endothelial cells, triggering robust inflammatory cascades. Animal models have demonstrated that transplanting the gut microbiota of aged mice into young, germ-free mice is sufficient to induce systemic inflammaging and subsequent neuroinflammation, confirming the causal role of the microbiome in establishing the peripheral inflammatory tone 12. Furthermore, specific bacterial taxa, including Staphylococcus aureus and Campylobacter jejuni, have been shown to directly activate nociceptors and vagal afferents, further entrenching the link between gut flora and central neurological responses 13.
Organ-Brain Axes and Systemic Resilience
Neurodegeneration is increasingly recognized as a multi-organ system failure. Mounting evidence indicates that pathology often begins outside the central nervous system, sometimes decades before the manifestation of classical motor or cognitive deficits 1. These varied organ-brain interactions function as distinct vulnerabilities where localized inflammation or protein misfolding can initiate systemic decline.
The lung-brain axis, for example, links chronic hypoxia, particulate matter exposure, and pulmonary inflammation to the acceleration of central protein misfolding 1. The musculoskeletal system contributes via the bone-brain axis, where myokines and osteokines modulate systemic resilience and mitochondrial health; the deterioration of this axis with age removes vital neuroprotective signals 1. Similarly, the endocrine-brain and reproductive axes dictate systemic metabolic and hormonal rhythms, and their age-related decline strongly influences the susceptibility of the central nervous system to inflammatory damage 1.
More specialized pathways, such as the bladder-brain and larynx-brain axes, highlight the vulnerability of autonomic and peripheral nerves to direct proteinopathic invasion. The abnormal accumulation of alpha-synuclein frequently occurs in the pelvic and pudendal nerves supplying the bladder, as well as the vagal branches innervating the larynx 1. This localized peripheral protein aggregation causes early autonomic symptoms, such as urinary dysfunction and dysphonia, and serves as a peripheral reservoir from which misfolded proteins may propagate in a prion-like manner toward the brainstem 1. Chronic pain also acts as a profound systemic stressor; peripheral pain pathways maintain central microglial activation through the continuous release of tumor necrosis factor-alpha and interleukin-1 beta, driving a feedback loop of central sensitization and neuroinflammation 14.
Epidemiological Modifiers of Inflammaging
The impact of systemic inflammation on neurodegenerative risk is not uniform; it is heavily modulated by demographic factors, genetic predispositions, and the totality of environmental exposures across a lifespan.
Systemic Immune-Inflammation Index
Global epidemiological studies utilizing comprehensive health databases, such as the National Health and Nutrition Examination Survey, have successfully quantified the link between peripheral inflammation and disease risk 1516. Metrics like the Systemic Immune-Inflammation Index, which integrates peripheral platelet, neutrophil, and lymphocyte counts, function as objective proxies for the host's overall inflammatory-immune balance 1617.
Cross-sectional analyses of over 54,000 adults have demonstrated a significant, positive, and non-linear dose-response relationship between elevated Systemic Immune-Inflammation Index levels and the prevalence of Parkinson's disease 1617. Data indicates a notable escalation in risk tied to the severity of peripheral inflammation; individuals in the highest quartile of systemic inflammation exhibit a 2.49-fold increased risk of developing Parkinson's disease (Odds Ratio: 2.49, 95% CI: 1.69 - 3.77) compared to those in the lowest quartile, while those in the third quartile face an 82% elevated risk (Odds Ratio: 1.82, 95% CI: 1.20 - 2.82) 16. This epidemiological evidence firmly positions peripheral immune activation as a robust, independent predictor of neurodegenerative vulnerability, distinct from primary central nervous system pathology. Similar blood-based indices, including the neutrophil-to-lymphocyte ratio and the systemic inflammation response index, have independently corroborated this association, indicating that higher baseline systemic inflammation correlates strongly with cognitive decline and motor dysfunction 1518.
Population-Specific Profiles and Genetic Modifiers
The expression and consequences of inflammaging are highly population-specific and shaped by the exposome - the cumulative burden of environmental, lifestyle, and infectious exposures. Studies evaluating diverse global populations reveal that classic markers of inflammaging, which align closely with chronological aging in industrialized nations, frequently fail to replicate in indigenous or non-industrialized populations 19. For instance, immune aging in populations with high endemic rates of intestinal parasites is governed by different cytokine patterns, suggesting that inflammaging is not a universal biological inevitability but a specific byproduct of industrialized lifestyles and environments 19.
In populations of African ancestry, specific inflammatory signatures act as primary drivers of neurodegeneration. Systematic reviews highlight that interleukin-6 shows significantly stronger correlations with Alzheimer's disease and cognitive decline in African populations compared to Caucasian cohorts 2021. Furthermore, unique genetic variants profoundly influence this inflammatory trajectory. Specific polymorphisms in the Triggering Receptor Expressed on Myeloid Cells 2 (TREM2), along with distinct Fc fragment of IgG receptor genotypes, uniquely alter Alzheimer's-related neuroinflammation in individuals of African descent 202122. Despite these specific vulnerabilities, research regarding the intersection of neurodegeneration and systemic trauma remains geographically skewed. Notably, there is a severe deficit in research concerning chronic traumatic encephalopathy within African populations, where repeated head injuries in contact sports likely intersect with these unique inflammatory and genetic baselines to produce unrecognized neurodegenerative burdens 23.
Pathways of Peripheral-to-Central Signaling
The central nervous system was historically viewed as an immune-privileged site, shielded from the peripheral circulation. It is now understood that peripheral inflammation actively communicates with the brain through structural and neural pathways, effectively translating a systemic state into localized neuroinflammation.
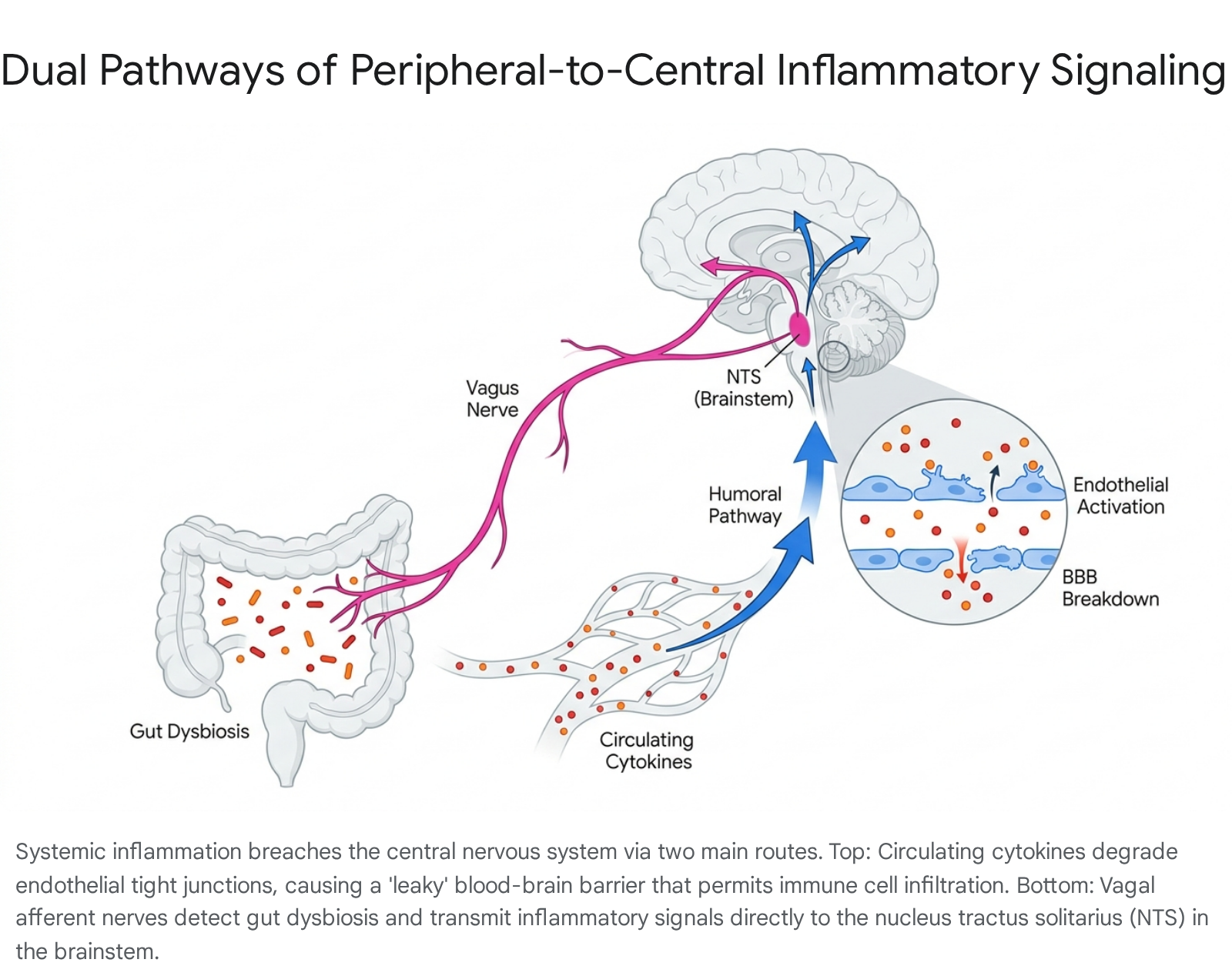
Blood-Brain Barrier Endothelial Activation
The blood-brain barrier regulates the molecular and cellular exchange between the systemic circulation and the brain parenchyma, maintaining the strict homeostatic environment required for neuronal function. The barrier consists of highly specialized endothelial cells, pericytes, and astrocytic endfeet 1124.
During states of peripheral inflammaging, elevated circulating cytokines (such as tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-6) and microbial byproducts (such as lipopolysaccharides) directly interact with receptors on the luminal surface of brain endothelial cells 5. This interaction activates intracellular signaling cascades, most prominently those mediated by the transcription factor Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-κB) 5. The activation of NF-κB triggers a profound phenotypic shift in the endothelium, leading to two critical structural compromises: 1. Tight Junction Degradation: The transcription and assembly of vital tight junction proteins, particularly claudin-5 and occludin, are heavily downregulated and degraded, resulting in increased paracellular permeability 511. 2. Upregulation of Adhesion Molecules: The activated endothelium upregulates the expression of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 5.
This endothelial activation creates a compromised, "leaky" barrier. Beyond physical breakdown, chronic inflammation induces the downregulation of glucose transporter 1, reducing glucose availability in the brain, and alters amino acid transport, fundamentally disrupting neurotransmitter balance 5. The upregulated adhesion molecules facilitate the transendothelial migration of peripheral immune cells - such as monocytes, T-cells, and B-cells - and neurotoxic plasma proteins directly into the brain parenchyma 2526. Once inside the central nervous system, infiltrating monocytes release additional chemokines, such as CCL2, and interact via direct cell-to-cell contact with resident glia 2425. This amplifies the inflammatory cascade and demonstrates that severe neuroinflammation can be triggered entirely by signaling from the periphery, without a primary injury originating within the brain 24.
Vagus Nerve and Brainstem Transmission
Beyond humoral blood-brain barrier pathways, the peripheral immune system utilizes direct neural routes to communicate inflammatory states to the brain. The vagus nerve serves as the primary bidirectional conduit for the gut-brain axis 13. Vagal afferent fibers innervating the gastrointestinal and respiratory tracts express numerous cytokine receptors and Toll-like receptors, enabling them to directly sense the local inflammatory milieu, bacterial toxins, and gut microbiome metabolites 1327.
Upon detecting peripheral cytokines, vagal sensory neurons - whose cell bodies reside in the nodose ganglia - transmit action potentials to the brainstem, specifically terminating in the nucleus tractus solitarius and the adjacent area postrema 2528. The area postrema is a circumventricular organ that lacks a traditional blood-brain barrier, making its local neuronal populations exquisitely sensitive to circulating blood-borne factors 2829.
Within the nucleus tractus solitarius, the incoming vagal signals stimulate the release of the excitatory neurotransmitter glutamate 25. This glutamatergic signaling rapidly alters the distribution of neuronal activity across autonomic brainstem nuclei, notably the ventrolateral medulla 28. This neural relay transmits the peripheral inflammatory signal deep into the central nervous system, directly modulating the state of resident microglial cells. Chronic signaling via this pathway, particularly in states of persistent gut dysbiosis, sustains central neuroinflammation. Consequently, conditions like chronic gastrointestinal distress are mechanistically linked to the progressive neurodegeneration and autonomic dysfunction frequently observed in the early stages of Parkinson's disease 1325. Interestingly, interventions such as vagus nerve stimulation have been shown to promote anti-inflammatory microglial phenotypes via alpha-7 nicotinic acetylcholine receptor pathways, indicating that this neural axis can be leveraged for therapeutic immune modulation 30.
Microglial Dynamics in Neurodegeneration
Microglia, the resident parenchymal macrophages of the central nervous system, are the primary effectors of neuroinflammation. Under physiological conditions, homeostatic microglia continuously survey the brain environment, clearing cellular debris, remodeling synapses, and providing neurotrophic support. However, in the context of inflammaging and neurodegenerative proteinopathies, microglial function becomes fundamentally dysregulated, shifting from a protective role to a highly destructive one.
The Cause Versus Consequence Debate
Historically, microglial activation in Alzheimer's and Parkinson's diseases was viewed as a secondary consequence - a reactive, somewhat passive response to the accumulation of amyloid-beta plaques, tau neurofibrillary tangles, or alpha-synuclein inclusions 31. While transient microglial activation is protective and actively aids in the phagocytic clearance of these misfolded proteins, recent genomic and transcriptomic evidence has inverted the traditional paradigm, positioning microglial dysfunction as a primary, causal driver of neurodegeneration 3132.
The modern consensus indicates a bidirectional causal relationship. Misfolded proteins and peripheral inflammatory signals initially trigger microglial activation, but sustained, chronic activation rapidly overwhelms the cells' protective mechanisms, inducing a profound neurotoxic state 3233. Overactivated microglia release high concentrations of reactive oxygen species and pro-inflammatory cytokines, including interleukin-1 beta, tumor necrosis factor-alpha, and interleukin-6 343536. The release of interleukin-1 beta is particularly deleterious, as it activates NLRP3 inflammasomes and alters microglial metabolic pathways, promoting further inflammatory reprogramming 36. This toxic microenvironment directly damages surrounding neurons, exacerbates protein misfolding, and triggers further glial activation, creating a self-perpetuating, feed-forward loop of chronic neuroinflammation that drives disease progression independently of the original protein aggregate trigger 3337.
Microglial Senescence and the Disease-Associated Phenotype
The simplistic categorization of microglia into "M1" (pro-inflammatory) and "M2" (anti-inflammatory) states is increasingly recognized as insufficient to describe the complex, overlapping transcriptomic signatures observed in neurodegeneration 3435. Advanced single-cell RNA sequencing has identified distinct subpopulations, most notably Disease-Associated Microglia and senescent microglia, which exhibit highly specialized and often deleterious functional profiles 838.
During aging and in the presence of continuous pathology, microglia gradually lose their homeostatic properties and adopt a senescent or dystrophic phenotype 3938. Morphologically, senescent microglia exhibit process de-ramification, abnormal swelling, cytorrhexis (fragmentation), vacuolization, and severe iron and ferritin accumulation 839. At the molecular level, these cells are characterized by elevated markers of cell cycle arrest, such as p16INK4a and p21CIP1, alongside profound dysregulation of cholesterol metabolism 10. Functionally, these cells suffer a profound loss of phagocytic capacity, rendering them unable to clear accumulating amyloid-beta or alpha-synuclein debris 3539.
Simultaneously, senescent microglia secrete a highly toxic senescence-associated secretory phenotype, heavily enriched in interleukin-6 and interleukin-8, driven by the activation of p38/mitogen-activated protein kinase and NF-κB pathways 8. The accumulation of these dysfunctional cells is regionally specific; senescent microglia are found in high concentrations in the hippocampus of Alzheimer's patients and the substantia nigra of Parkinson's patients, directly correlating with the specific neuroanatomical areas of maximum neuronal loss 839.
Crucial genetic risk factors regulate this transition. The Apolipoprotein E4 allele, the strongest genetic risk factor for late-onset Alzheimer's disease, actively suppresses protective microglial responses. It alters microglial lipid metabolism, induces mitochondrial dysfunction, and forces microglia into a stress-related transcriptomic signature, severely impairing their phagocytic efficacy 3540. Conversely, Triggering Receptor Expressed on Myeloid Cells 2 - a microglial surface receptor essential for damage sensing and phagocytosis - plays a complex, dual role. While functional TREM2 is required to clear plaques, altered TREM2 signaling is heavily implicated in facilitating the transition of microglia into the pro-inflammatory Disease-Associated Microglia phenotype 840.
Complement-Mediated Synaptic Pruning
One of the most destructive physiological consequences of microglial overactivation is the aberrant elimination of neuronal synapses, a process that underlies the severe cognitive and motor deficits defining Alzheimer's and Parkinson's diseases. During healthy embryonic and early postnatal development, microglia are essential for refining neural circuits, actively pruning weak or excess synapses using the classical complement cascade 4142.
In neurodegenerative disease, this developmental pruning mechanism is pathologically reactivated. Inflammaging and localized neurotoxicity cause an abnormal upregulation of complement proteins, particularly C1q, C3, and C4, which coat or "opsonize" functional, healthy synapses 414543. Microglia, expressing the Complement Receptor 3, recognize these C3-tagged synapses and actively engulf them via phagocytosis 43. Additionally, phagocytic receptors such as MEGF10 and MERTK on microglia, alongside reactive astrocytes upregulating the ABCA1 pathway, further contribute to this coordinated synaptic destruction 43.
In Alzheimer's models, C1q binds directly to fibrillar amyloid-beta and tau proteins, initiating the classical complement cascade and erroneously directing microglia to destroy adjacent, otherwise healthy synapses 44. The receptor interaction between C3a and its receptor (C3aR) further mediates detrimental crosstalk among neurons, microglia, and astrocytes 42. Crucially, this excessive complement-mediated synaptic pruning occurs early in the disease process, often preceding widespread neuronal death or the formation of visible plaques, establishing it as a primary mechanism of early cognitive decline 4344.
Divergent Signatures in Alzheimer's and Parkinson's
While Alzheimer's and Parkinson's diseases share the foundational mechanism of systemic inflammaging driving central microglial dysfunction, the diseases exhibit distinct pathophysiological profiles, spatial distributions, and primary proteinopathies.
Alzheimer's Disease Pathogenesis
Alzheimer's disease is primarily characterized by the accumulation of extracellular amyloid-beta plaques and intracellular hyperphosphorylated tau tangles 3335. The pathology predominantly targets the hippocampus and cortical gray matter, manifesting as severe deficits in memory and executive function 3739. In the context of neuroinflammation, Alzheimer's models frequently exhibit unique microglial phenotypes, such as "Dark Microglia," which are defined by markers of severe oxidative stress, condensed nucleoplasm, and mitochondrial alterations 45. Furthermore, Alzheimer's pathology is uniquely associated with the senescence of oligodendrocyte progenitor cells, and the neuroinflammatory response is closely tracked via cerebrospinal fluid biomarkers including Aβ42, specific tau isoforms, and neurofilament light chain, alongside plasma markers like soluble TREM2 and C1q 45.
Parkinson's Disease Pathogenesis
Conversely, Parkinson's disease is defined by the intracellular accumulation of misfolded alpha-synuclein proteins, forming Lewy bodies 3539. This pathology is highly localized to the dopaminergic neurons of the Substantia Nigra pars compacta and the midbrain, leading to the characteristic motor symptoms of rigidity, bradykinesia, and tremor 3739. Microglial dysfunction in Parkinson's is marked by a severe, age-related accumulation of senescent microglia in the substantia nigra, which exhibit a profound inability to clear alpha-synuclein aggregates 839. The peripheral prodrome of Parkinson's is notably distinct; patients frequently experience decades-long autonomic and systemic precursors, including severe constipation, gut dysbiosis, and REM sleep behavior disorder, highlighting the strong influence of the gut-brain axis 1. Systemic inflammation in Parkinson's is frequently tracked via elevated complete blood count-derived metrics, such as the Systemic Immune-Inflammation Index and the Neutrophil-to-Lymphocyte Ratio 1618.
| Pathological Feature | Alzheimer's Disease (AD) | Parkinson's Disease (PD) |
|---|---|---|
| Primary Proteinopathy | Amyloid-beta (Aβ) plaques and hyperphosphorylated Tau tangles 3335. | Alpha-synuclein inclusions (Lewy bodies) 3539. |
| Primary Brain Region | Hippocampus and cortical gray matter (memory and executive function) 3739. | Substantia Nigra pars compacta and midbrain (motor control) 3739. |
| Microglial Characteristics | "Dark Microglia" (oxidative stress); senescent oligodendrocyte progenitors 4546. | Age-related accumulation in the Substantia Nigra; severe iron/ferritin accumulation 83945. |
| Systemic Prodrome | Post-operative cognitive dysfunction, localized systemic inflammation 14. | Decades-long autonomic prodrome: Constipation, dysbiosis, sleep behavior disorders 1. |
| Shared Inflammatory Factors | Elevated circulating IL-1β, IL-6, TNF-α; compromised blood-brain barrier 45. | Elevated circulating IL-1β, IL-6, TNF-α; compromised blood-brain barrier 45. |
| Clinical Biomarkers | CSF Aβ42, Tau, NfL; plasma sTREM2, YKL-40, C1q 45. | High Systemic Immune-Inflammation Index, Neutrophil-to-Lymphocyte ratio 1618. |
The Therapeutic Pipeline Targeting Neuroinflammation
The recognition of neuroinflammation and the blood-brain barrier as critical obstacles to therapeutic success has catalyzed a new generation of disease-modifying drug candidates. As the industry moves into 2026, the clinical pipeline features advanced programs designed specifically to cross the blood-brain barrier, modulate microglial behavior, and clear misfolded proteins before the feed-forward cycle of neurodegeneration causes irreversible damage.
Blood-Brain Barrier Penetrant Platforms
Traditional monoclonal antibodies and large-molecule therapeutics suffer from exceptionally low blood-brain barrier penetrance, severely limiting their concentration and efficacy in the central nervous system. Biopharmaceutical companies have developed novel delivery platforms to overcome this structural hurdle. Denali Therapeutics utilizes a proprietary TransportVehicle platform, which engineers large molecules - including enzymes, oligonucleotides, and antibodies - to bind to transcytosis receptors, such as the transferrin receptor, on the blood-brain barrier 4748. This allows the therapeutic to be actively ferried across the endothelium and into the brain parenchyma, achieving broad biodistribution.
Denali's pipeline includes multiple TransportVehicle-enabled candidates advancing through clinical trials in 2026. DNL628 is an Oligonucleotide TransportVehicle designed to target the MAPT gene and reduce tau protein synthesis in early Alzheimer's disease; the first patient was dosed in a Phase 1b study in March 2026, with data expected in early 2027 49. Concurrently, DNL921 represents an Antibody TransportVehicle targeting amyloid-beta. Preclinical data published in Science indicates that the TransportVehicle platform improves brain distribution and bypasses amyloid-laden large vessels by traveling through smaller capillaries 4850. This specialized delivery significantly reduces the risk of amyloid-related imaging abnormalities, a severe vascular side effect that limits the utility of first-generation anti-amyloid therapies 48. Denali is also preparing for the potential commercial launch of tividenofusp alfa, an enzyme replacement therapy for Hunter syndrome, pending a regulatory decision expected in April 2026 .
Kinase Inhibitors and Anti-Inflammatory Modulators
Beyond specialized delivery platforms, therapeutics are increasingly targeting the intracellular signaling cascades that fuel microglial senescence and the inflammatory response.
For Parkinson's disease, the pipeline is highly diversified. BIIB122 (also known as DNL151), a central nervous system-penetrant small-molecule inhibitor of Leucine-Rich Repeat Kinase 2 developed by Biogen and Denali, completed enrollment for its global Phase 2b LUMA study, with a highly anticipated clinical readout expected in mid-2026 51. Because mutations and overactivity of Leucine-Rich Repeat Kinase 2 are heavily implicated in lysosomal dysfunction and microglial hyperactivation in Parkinson's disease, inhibiting this kinase aims to restore cellular homeostasis and slow overall disease progression 5152.
Additionally, Biohaven is advancing BHV-8000, a highly selective, brain-penetrant TYK2/JAK1 inhibitor, which is currently progressing through a pivotal Phase 2/3 trial for early Parkinson's disease 59. By directly inhibiting the TYK2 and JAK1 pathways, BHV-8000 suppresses the transmission of inflammatory cytokine signals between cells, directly targeting the neuroinflammation loop that drives disease worsening 53. Early clinical experience demonstrated that BHV-8000 effectively enters the brain and significantly reduces blood biomarkers of systemic inflammation with a favorable safety profile 59.
Monoclonal Antibodies and Microglial Depletion
Therapeutic approaches are also attempting to clear toxic protein aggregates directly or fundamentally "reset" the microglial population. In the Alzheimer's space, UCB's investigational anti-tau monoclonal antibody, bepranemab, recently completed the Phase 2a TOGETHER trial 54. While the trial did not meet its primary clinical endpoint regarding overall cognitive decline across the broad patient cohort, bepranemab successfully reduced tau accumulation by 33% to 58% relative to placebo - marking the first time a tau-directed therapy demonstrated clinical target engagement 54. Furthermore, a predefined subgroup analysis revealed significant clinical benefit, halving the rate of decline on the ADAS-Cog scale in patients with low baseline tau burden who were non-carriers of the APOE4 allele 5455. Despite these localized successes, Roche and Genentech returned the development rights for bepranemab to UCB following the trial's conclusion 55.
For Parkinson's disease, UCB recently completed a successful Phase 1 safety trial for UCB7853, an anti-alpha-synuclein antibody designed to prevent the extracellular spread of the toxic protein 5657. However, the broader alpha-synuclein targeted space has faced setbacks, notably with UCB and Neuropore halting the development of minzasolmin after the Phase 2 ORCHESTRA study failed to meet its primary and secondary endpoints 5765.
Finally, highly experimental approaches aim to address the senescent microglial population directly. Colony-Stimulating Factor 1 Receptor inhibitors, such as PLX3397 and PLX5622, are being explored extensively in preclinical models as a method to safely and selectively deplete senescent, toxic microglia 5859. Following targeted depletion, the cessation of the inhibitor allows the brain to repopulate with naive, functional microglia derived from local progenitors, effectively "resetting" the central immune environment. While early generation inhibitors exhibit poor blood-brain barrier penetrance and associated liver toxicity, advanced cellular therapies utilizing targeted Colony-Stimulating Factor 1 Receptor mechanisms are under active, highly promising investigation for both Alzheimer's and Parkinson's disease 5860.
| Therapeutic Candidate | Sponsor / Developer | Target / Mechanism | Indication | Current Clinical Status (2026) |
|---|---|---|---|---|
| BIIB122 (DNL151) | Biogen / Denali | Small molecule LRRK2 inhibitor | Early-stage Parkinson's disease | Phase 2b (LUMA) fully enrolled; clinical readout expected mid-2026 . |
| BHV-8000 | Biohaven | Brain-penetrant TYK2/JAK1 inhibitor | Early-stage Parkinson's disease | Pivotal Phase 2/3 currently enrolling and advancing . |
| Bepranemab | UCB | Monoclonal anti-tau antibody | Mild to moderate Alzheimer's disease | Phase 2a completed; reduced tau but missed primary endpoint; rights returned 5455. |
| UCB7853 | UCB | Anti-alpha-synuclein antibody | Parkinson's disease | Phase 1 completed successfully; demonstrated safety and target engagement 5657. |
| DNL628 (OTV:MAPT) | Denali | TV-enabled oligonucleotide (Tau) | Alzheimer's disease | Phase 1b ongoing; first patient dosed March 2026, data expected 1H 2027 49. |
| DNL921 (ATV:Abeta) | Denali | TV-enabled antibody (Amyloid-beta) | Alzheimer's disease | IND-enabling stage; Phase 1 start-up activities in progress 48. |
| Minzasolmin | UCB / Neuropore | Alpha-synuclein inhibitor | Parkinson's disease | Phase 2 (ORCHESTRA) failed primary/secondary endpoints; development halted 5765. |
Conclusion
The characterization of the inflammaging-neurodegeneration axis has fundamentally reshaped the scientific understanding of Alzheimer's and Parkinson's diseases. Neurodegeneration is no longer viewed as an isolated, localized pathology of the brain, but rather as the complex endpoint of a systemic, lifelong cascade involving gut dysbiosis, peripheral cellular senescence, and widespread barrier breakdown. Chronic systemic inflammation physically breaches the central nervous system via both humoral endothelial pathways and direct neural vagal transmission. This relentless peripheral signaling forcefully shifts resident microglia from a homeostatic, neuroprotective role into a senescent, pro-inflammatory, and highly destructive phenotype. The subsequent complement-mediated pruning of healthy synapses by these dysfunctional microglia drives the early, devastating cognitive and motor declines that define these irreversible disorders.
As the field progresses through 2026, the therapeutic landscape closely reflects this systemic, multi-axis understanding. The integration of advanced blood-brain barrier transport technologies, highly specific kinase inhibitors targeting the inflammatory cascade, and precision medicine approaches tailored to diverse genetic and demographic populations offers a profound evolution in strategy. By intervening early in the inflammatory cascade and directly addressing the peripheral-to-central dialogue, modern neurotherapeutics aim not merely to manage late-stage symptoms, but to halt the fundamental biological processes driving brain aging and disease.