Stem cell exhaustion and restoration in aging
Somatic stem cells function as the biological foundation for tissue homeostasis and regenerative repair throughout the human lifespan. These localized populations possess the dual capacity to self-renew, thereby maintaining the stem cell reservoir, and to differentiate into specialized cell types that replenish tissues lost to injury, daily wear, or disease 12. However, the regenerative capacity of these stem cell pools undergoes a profound and progressive decline with advancing age, a phenomenon collectively termed stem cell exhaustion 13.
The depletion of functional stem cell reserves manifests biologically as an inability to maintain tissue architecture, leading directly to the onset of age-related morbidities. These pathologies include immunosenescence, sarcopenia, osteopenia, neurodegeneration, and impaired wound healing 34. Stem cell exhaustion is not driven by a singular biological failure but is instead the culmination of complex, interconnected dysfunctions. These dysfunctions are broadly categorized into intrinsic cellular defects - such as genomic instability, epigenetic drift, and metabolic failure - and extrinsic disruptions originating from the stem cell microenvironment, or niche 145.
In recent years, the understanding of stem cell aging has shifted from a model of inevitable cellular decay to a paradigm of modifiable biological circuits. Researchers have demonstrated that the functional decline of adult stem cells can be delayed, and in some cases reversed, through targeted interventions 56. Two highly promising therapeutic avenues have emerged to counteract stem cell exhaustion: the clearance of senescent cells from the niche using senolytics to restore the extrinsic microenvironment, and the use of partial epigenetic reprogramming via Yamanaka factors to restore the intrinsic youthfulness of the stem cells themselves 789.
Mechanisms of Intrinsic Stem Cell Decline
While adult stem cells are relatively protected from certain aging stressors compared to their rapidly dividing differentiated progeny, they remain susceptible to the progressive accumulation of molecular damage over decades 1. Intrinsic aging refers to the cell-autonomous alterations that degrade a stem cell's ability to maintain quiescence, execute symmetric or asymmetric division, and commit to appropriate differentiation lineages 49.
Genomic Instability and DNA Damage
The accumulation of DNA damage is a primary driver of intrinsic stem cell exhaustion. Throughout an organism's lifespan, stem cells are exposed to endogenous metabolic byproducts, such as reactive oxygen species (ROS), as well as exogenous stressors that cause double-strand breaks and errors in DNA synthesis 29. In youthful tissues, DNA repair mechanisms, such as non-homologous end joining and homologous recombination, efficiently resolve these lesions. However, the efficacy of these repair pathways declines with age. Mouse models with mutations in DNA repair, such as a hypomorphic allele in DNA ligase IV (LigIV), exhibit severe deficits in stem cell function that mimic natural aging 210.
Persistent DNA damage triggers intrinsic stress responses that force the stem cell out of its normal functional state. The activation of DNA damage-repair responses stimulates cell cycle checkpoints, leading either to senescence mediated by the p16INK4a pathway, or apoptosis mediated by the p53-PUMA pathway 9. Additionally, the CD53-p21 signaling axis induces cell cycle arrest 9. The continuous activation of these tumor-suppressor pathways ultimately depletes the active stem cell pool. The surviving stem cells are subsequently forced to undergo compensatory hyperproliferation, which accelerates telomere attrition and further destabilizes the genome in a destructive feedback loop 39. Furthermore, the deletion of critical transcription factors, such as the FoxO family (Foxo1, Foxo3a, and Foxo4), which normally regulate oxidative stress responses, results in a significant decrease in stem and progenitor cell self-renewal 2.
Epigenetic Drift and Transcriptional Dysregulation
Beyond structural damage to the DNA sequence, the aging of stem cells is characterized by profound changes in the epigenome. Epigenetic drift involves the gradual loss of precise DNA methylation patterns, the dysregulation of histone modifications, and the deterioration of three-dimensional chromatin architecture 1011. These alterations destabilize the tightly controlled transcriptional programs required to maintain stem cell quiescence and regulate lineage commitment 10.
In youthful stem cells, maintaining a state of shallow quiescence is pivotal for preserving stemness, minimizing metabolic stress, and protecting genomic integrity 9. Epigenetic dysregulation frequently forces aging stem cells to exit quiescence prematurely or impairs their ability to re-enter it following an injury response. Alterations in enhancer methylation and chromatin looping can lead to aberrant gene expression profiles that skew the differentiation potential of the stem cell. Interestingly, differentially methylated genes in aged stem cells are often not highly expressed in the stem cells themselves but become anomalously active as the cells commit to downstream progenitor states, highlighting how epigenetic memory drives delayed functional deficits 11.
Metabolic Dysfunction and Proteostasis Failure
Metabolic inflexibility and the loss of protein homeostasis (proteostasis) represent the third pillar of intrinsic stem cell decline. Aging disrupts critical nutrient- and energy-sensing pathways, notably the mammalian target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) signaling networks 10. Dysregulation of these pathways leads to impaired autophagy, which is the cellular mechanism responsible for degrading and recycling damaged organelles and misfolded proteins 1012.
In aging stem cells, defective autophagy leads to the accumulation of toxic protein aggregates and dysfunctional mitochondria. Mitochondrial dysfunction, in turn, exacerbates the production of intracellular ROS, creating a detrimental feedback loop that inflicts further damage on the genome and epigenome 1011. Recent research has identified specific proteins, such as AP2A1, that play a significant role in cellular aging. Lowering AP2A1 levels via targeted compounds like IU1 - which inhibits the deubiquitinating enzyme USP14 - has been shown to enhance proteasome activity and autophagy, accelerating the removal of toxic cellular waste and reversing aspects of cellular aging 12. The inability to maintain a pristine proteome and a functional mitochondrial network otherwise forces stem cells into metabolic stress, severely compromising their regenerative output upon activation 1013.
Extrinsic Deterioration of the Stem Cell Niche
Stem cells do not exist in isolation; they reside within highly specialized microenvironments known as niches. The niche is a complex biological architecture composed of extracellular matrix (ECM) components, vascular networks, stromal support cells, and resident immune cells 141516. This environment provides crucial biochemical and physical signals that dictate stem cell behavior, regulating the delicate balance between quiescence, proliferation, and differentiation 2.
In a youthful state, the stem cell niche provides optimal biochemical and structural support, featuring healthy, elastic extracellular matrix fibers, anti-inflammatory macrophages, and precise gradients of growth factors that maintain stem cells in a healthy, quiescent state. However, during aging, the accumulation of senescent cells transforms the niche into a pro-inflammatory, fibrotic environment, actively suppressing stem cell regeneration. Senescent cells enter an irreversible state of cell cycle arrest but remain metabolically active, secreting a highly toxic mixture of pro-inflammatory cytokines, chemokines, matrix metalloproteinases, and growth factors 181718. This profile is known as the senescence-associated secretory phenotype (SASP).
Cellular Senescence and Inflammaging
The SASP transforms the formerly supportive niche into a highly inflammatory zone, a state frequently referred to as "inflammaging" 61519. The chronic, low-grade inflammation generated by these senescent cells directly suppresses stem cell activity by altering the local cytokine milieu 1518. Furthermore, the SASP induces a "bystander effect," where secreted factors and extracellular vesicles (EVs) trigger senescence in neighboring, previously healthy cells, amplifying the dysfunction exponentially throughout the tissue 7.
Structural and Extracellular Matrix Alterations
The deterioration of the niche is not strictly biochemical; it is heavily mechanical. SASP-induced dysregulation of tissue remodeling leads to profound structural changes in the extracellular matrix. Elevated collagen cross-linking and a shift from cellular tissue to fibrotic scar tissue physically stiffen the microenvironment 1620. This mechanical stiffness impedes the physical mobility, engraftment, and volumetric expansion of stem cells required for effective tissue repair. Consequently, even if a stem cell retains intrinsic youthfulness, it cannot effectively navigate or function within a stiffened, fibrotic matrix 20.
Pathological Profiles Across Tissue Compartments
While the broad mechanisms of intrinsic damage and niche deterioration apply universally across the organism, the specific biological manifestations of stem cell exhaustion vary significantly across different organ systems. The unique physiological demands, cellular compositions, and regenerative turnover rates of each tissue dictate how stem cell decline presents pathologically.
Hematopoietic Stem Cells (HSCs)
Hematopoietic stem cells, residing in the bone marrow, are responsible for the lifelong production of the entire blood and immune system. As humans age, the overall number of phenotypically defined HSCs paradoxically increases, but their functional regenerative capacity drastically drops 1114.
The hallmark of HSC aging is myeloid skewing. Youthful HSCs maintain a balanced output of both myeloid cells (granulocytes, monocytes, macrophages) and lymphoid cells (B-cells, T-cells). In aged HSCs, differentiation potential heavily biases toward the myeloid lineage at the expense of lymphopoiesis 2511. This skewing is a primary driver of immunosenescence, rendering older adults highly susceptible to novel infections and reducing adaptive immunity, while simultaneously flooding the systemic circulation with pro-inflammatory myeloid cells that exacerbate organismal aging 515.
At the molecular level, human HSC aging is characterized by the accumulation of a senescent cell cluster known as MPP2A. This specific cellular subset is defined by severe telomere attrition, persistent cell cycle arrest, and a significant upregulation of the P53-P21CIP1 pathway, which notably contrasts with murine models that primarily rely on the p16INK4a pathway 21. Additionally, aged human HSCs exhibit aberrantly elevated activity of the small RhoGTPase Cdc42, a protein responsible for cellular organization. Elevated Cdc42 disrupts cell polarity, which delays the initiation of cell division and impairs asymmetric self-renewal 22.
The bone marrow niche also deteriorates severely. Alterations in perivascular mesenchymal stromal cells (MSCs) and degeneration of sympathetic adrenergic nerve signals disrupt the homing environment 1423. Furthermore, megakaryocytes - which regulate HSC quiescence via factors like CXCL4 - expand in number and alter their spatial proximity to HSCs 14. Collectively, these niche alterations selectively support functionally impaired, myeloid-biased HSC clones, setting the stage for clonal hematopoiesis and an elevated risk of hematological malignancies 1423. In specific pathological contexts, such as Sickle Cell Disease (SCD), the bone marrow niche experiences accelerated damage, resulting in profound HSC dysfunction and premature epigenetic aging 2425.
Mesenchymal Stromal Cells (MSCs)
Mesenchymal stem/stromal cells are multipotent progenitors found in various tissues, most prominently the bone marrow and adipose tissue. Unlike HSCs, which increase in absolute number, MSCs suffer a catastrophic quantitative depletion with age, decreasing 100- to 1,000-fold in frequency between the third and sixth decades of human life 26.
The primary physiological role of MSCs extends beyond direct cellular replacement; they are vital orchestrators of tissue repair, secreting regenerative growth factors, anti-inflammatory mediators, and extracellular vesicles that resolve chronic inflammation 2629. The precipitous loss of MSCs directly correlates with the exponential rise in degenerative diseases, such as severe osteoarthritis requiring joint replacement, and cardiovascular disease 26. Furthermore, intrinsic epigenetic changes cause the remaining aged MSCs to alter their differentiation trajectories, favoring adipogenesis (fat formation) over osteogenesis (bone formation) 1727. This lineage shift contributes directly to age-related osteopenia, osteoporosis, and increased, dysfunctional bone marrow adiposity 61727.
Muscle Satellite Cells (MuSCs)
Skeletal muscle regeneration relies entirely on resident muscle stem cells, known as satellite cells (MuSCs). In young, healthy muscle, satellite cells remain tightly quiescent beneath the basal lamina of muscle fibers, activating rapidly in response to injury to proliferate and fuse into new myofibers 1328.
With advancing age, the satellite cell pool becomes depleted and functionally exhausted, a primary contributor to sarcopenia - the progressive loss of muscle mass, strength, and metabolic capacity 31628. This exhaustion is heavily driven by intrinsic epigenetic silencing. For instance, the SPRY1 gene is a known regulator of MuSC quiescence; aging leads to increased methylation of SPRY1, which inhibits the replenishment of the stem cell pool 19.
Extrinsic niche deterioration is equally devastating to muscle regeneration. Fibro-adipogenic progenitors (FAPs), an essential support cell in the muscle niche, become senescent with age. Senescent FAPs secrete factors that impair satellite cell function and drive muscle fibrosis 1928. Additionally, systemic changes - such as reduced levels of circulating insulin-like growth factor-1 (IGF-1) and diminished macrophage-derived signaling like the cytokine CXCL10 - leave aged satellite cells unable to activate and divide efficiently 1319. The immune modulator mesencephalic astrocyte-derived neurotrophic factor (MANF) is also impaired 19. The suppressive nature of the aged niche is dominant; studies have shown that when youthful, highly functional satellite cells are cultured on extracellular matrix derived from aged mice, they completely fail to proliferate due to elevated collagen protein levels and mechanical stiffness 20. Rescuing intrinsic defects via an inducible FGF receptor 1 (FGFR1) must be combined with anti-fibrotic treatments to achieve meaningful muscle mass restoration 20.
Intestinal Stem Cells (ISCs)
The intestinal epithelium is one of the most rapidly renewing tissues in the mammalian body, driven by highly active intestinal stem cells (ISCs) located at the base of the crypts of Lieberkühn 2930. Aging drastically impairs ISC activity, leading to reduced crypt density, shorter intestinal villi, and a compromised intestinal barrier. This "leaky gut" syndrome allows the systemic leakage of microbiome-derived inflammatory factors, fueling whole-body inflammaging 330.
Recent investigations have uncovered precise molecular cross-talk between ISCs and their niche support cells, the Paneth cells. With age, high activity of the mTORC1 pathway in Paneth cells inhibits the peroxisome proliferator-activated receptor alpha (PPAR-alpha) 2934. This metabolic shift induces the production of Notum, an extracellular Wnt inhibitor 2934. Because Wnt signaling is strictly required to maintain ISC stemness and drive proliferation, the secretion of Notum by the aged niche actively suppresses intestinal regeneration 29. Furthermore, older ISCs lose their metabolic flexibility, exhibiting a significantly reduced capacity to adapt to changing nutrient statuses, which permanently alters their physical size and restricts their differentiation trajectories 3132.
Neural Stem Cells (NSCs)
In the adult human brain, neurogenesis is primarily restricted to specific niches, such as the subventricular zone and the dentate gyrus of the hippocampus 27. Neural stem cells (NSCs) in these regions maintain brain plasticity, support synaptic remodeling, and facilitate recovery from injury 37.
Aging severely restricts NSC function, contributing to cognitive decline and the progression of neurodegenerative diseases like Alzheimer's and Parkinson's 67. The accumulation of senescent cells within the central nervous system - including microglia, astrocytes, and endothelial cells - floods the neural niche with neuroinflammatory SASP factors 7. Senescent endothelial cells specifically compromise the integrity of the blood-brain barrier (BBB), increasing its permeability and allowing harmful peripheral immune cells and systemic inflammatory molecules to infiltrate the central nervous system 7. This breakdown in vascular integrity is strongly linked to white matter lesions and vascular cognitive impairment 7. Furthermore, factors secreted by aged bone marrow cells, such as cyclophilin A, can cross into the brain and directly contribute to synaptic loss and cognitive decline, demonstrating a systemic link between distant aging niches 37.
| Stem Cell Type | Primary Tissue Location | Dominant Intrinsic Aging Mechanisms | Key Niche & Extrinsic Disrupters | Major Pathological Consequences |
|---|---|---|---|---|
| Hematopoietic (HSCs) | Bone Marrow | Elevated Cdc42; telomere attrition; p53-p21CIP1 activation (MPP2A cluster). | Megakaryocyte expansion; loss of sympathetic signaling; MSC aging. | Myeloid skewing; immunosenescence; clonal hematopoiesis. |
| Mesenchymal (MSCs) | Bone Marrow, Adipose | 100- to 1000-fold numerical depletion; epigenetic shift toward adipogenesis. | Accumulation of SASP; systemic chronic inflammation. | Osteoporosis; delayed wound repair; increased frailty; CVD. |
| Muscle Satellite (MuSCs) | Skeletal Muscle | SPRY1 hypermethylation; loss of quiescence; diminished activation. | Senescent fibro-adipogenic progenitors (FAPs); stiffened collagen ECM. | Sarcopenia; failed regeneration after injury; muscle fibrosis. |
| Intestinal (ISCs) | Intestinal Crypts | Nutrient-sensing failure; enlarged physical cell size limiting differentiation. | Secretion of Wnt-inhibitor Notum by aged Paneth cells (mTORC1 hyperactive). | Blunted villi/crypts; nutrient malabsorption; systemic barrier leakage. |
| Neural (NSCs) | Hippocampus, SVZ | Reduced proliferative capacity; loss of intrinsic neurogenic potential. | Senescent glia/microglia; neuroinflammation; BBB breakdown. | Cognitive decline; impaired synaptic plasticity; neurodegeneration. |
Senotherapeutics for Niche Restoration
Because intrinsic stem cell exhaustion is heavily exacerbated by the toxic microenvironment, clearing senescent cells from the niche represents a highly viable strategy to restore regenerative capacity. Senotherapeutics are divided into two main categories: senolytics, which selectively induce apoptosis in senescent cells, and senomorphics, which suppress the secretion of the SASP without killing the cells 71718.
Pharmacological Senolytics and Senomorphics
Senescent cells evade natural immune clearance and resist apoptosis by upregulating pro-survival signaling networks, collectively termed senescent cell anti-apoptotic pathways (SCAPs) 7. These include the BCL-2 protein family, which governs mitochondrial apoptosis pathways, and the PI3K/AKT/mTOR signaling axis 730. Another critical survival mechanism is the interaction between the FOXO4 transcription factor and p53, which blocks p53-mediated apoptosis 18. Pharmacological senolytics are designed to transiently disrupt these specific SCAPs, triggering targeted cell death in senescent populations while sparing healthy cells 71833.
The most extensively studied senolytic intervention is the combination of dasatinib (a tyrosine kinase inhibitor that interferes with EFNB-dependent suppression of apoptosis) and quercetin (a natural flavonol that inhibits PI3K and serpins), collectively referred to as D+Q 183033. In aged mice, D+Q administration successfully clears senescent osteocytes and bone marrow cells. This clearance significantly improves the osteogenic capacity of aged BMSCs, resulting in increased bone volume and a reduction in frailty markers 1733. In the gastrointestinal tract, elderly mice treated with D+Q exhibited a marked decrease in p16 and p21 senescence markers and the physical restoration of intestinal villi and crypts, directly indicating an enhancement of ISC regenerative activity 3034. D+Q has also shown promise in early human pilot trials, notably reducing senescent-cell burden in the adipose tissue of diabetic kidney disease patients and lowering circulating SASP factors 40.
Targeted senolytics are also being deployed in specific pathological models and organ systems. Navitoclax (ABT-263), a potent BCL-2/BCL-xL inhibitor, has demonstrated remarkable efficacy in rescuing hematopoietic stem cell function in murine models of Sickle Cell Disease 2425. By clearing the accumulated senescent HSPCs, navitoclax restored hematopoietic repopulating activity 24. In the central nervous system, navitoclax has been utilized in models of global cerebral ischemia (GCI), where it cleared senescent cells, decreased SASP-mediated inflammation, and restored BBB function, facilitating neuronal survival post-injury 35. Similarly, the flavonoid fisetin decreases oxidative stress burden and downregulates NF-κB signaling, dampening SASP output and alleviating neuroinflammation 18.
In the clinical space, senolytics are increasingly shifting toward localized delivery to avoid systemic toxicity. UNITY Biotechnology's UBX1325, a senolytic BCL-xL inhibitor, is delivered directly into the eye for diabetic macular edema, showing promising early phase results 40. Dermatology is another localized frontier; Rubedo Life Sciences initiated a Phase 1 trial for RLS-1496, a topical GPX4-modulating senolytic aimed at clearing senescent cells associated with plaque psoriasis and skin aging 40.
Furthermore, existing FDA-approved drugs are being repurposed for their niche-clearing properties. Recent Japanese research identified canagliflozin, a common diabetes medication, as a potent activator of the longevity-associated enzyme AMPK. In murine models, canagliflozin treatment reduced senescence in adipose tissue and stimulated immune-mediated clearance of highly stubborn, immune-evading senescent cells (characterized by high PD-L1 and PD-1 expression), thereby prolonging lifespan and preserving physical strength 36.
Cellular Senolytics and Immune Modulation
While small-molecule senolytics are advancing through clinical trials, their systemic efficacy can be limited by off-target toxicity, the heterogeneous nature of the SASP, and tissue-specific resistance to apoptosis 740. To achieve higher precision and sustained efficacy, researchers are engineering cell-based senotherapies utilizing the immune system.
Natural Killer (NK) cells are the innate immune system's primary mechanism for surveying and clearing senescent cells. Because endogenous NK function declines significantly with age, the adoptive transfer of youthful or hyper-functional NK cells is being explored as an immunotherapeutic strategy to rejuvenate immunosenescence and actively eradicate SASP-producing cells across various tissue niches 37.
More targeted and aggressive approaches involve the use of Chimeric Antigen Receptor (CAR) T-cells. CAR T-cells can be engineered to recognize specific surface antigens unique to senescent populations. In recent breakthroughs, T-cells were engineered to target the urokinase-plasminogen activator receptor (uPAR), a surface antigen highly expressed on senescent cells during natural aging 38. In murine models, uPAR-targeted senolytic CAR T-cell therapy safely and effectively ablated senescent cells in vivo. When applied to the aging intestine, this cellular therapy cleared the aged niche, improved epithelial integrity, and significantly promoted the regenerative potential of exhausted ISCs, highlighting the future of precision niche restoration 38.
Partial Epigenetic Reprogramming: Reversing Intrinsic Aging
While senolytics address the extrinsic deterioration of the microenvironment by removing toxic cells, they cannot reverse the intrinsic epigenetic drift that has accumulated within the DNA of the surviving stem cells. To fundamentally reset the biological clock of somatic cells, the field of regenerative medicine has turned to partial epigenetic reprogramming 91845.
Principles of the Yamanaka Factors
In 2006, Kyoto University researcher Shinya Yamanaka demonstrated that the introduction of four specific transcription factors - Oct4, Sox2, Klf4, and c-Myc (collectively known as OSKM) - could completely reprogram a mature, terminally differentiated adult cell back into a pluripotent embryonic state, creating induced pluripotent stem cells (iPSCs) 454639. This Nobel Prize-winning discovery proved that the epigenetic changes driving cellular aging and specialization are entirely reversible.
However, inducing full pluripotency in vivo is lethal. If somatic cells are fully reprogrammed inside a living organism, they lose their functional tissue identity and rapidly form teratomas (complex tumors) 8918. To harness the rejuvenating power of OSKM without inducing cancer, scientists developed partial epigenetic reprogramming. By transiently and cyclically expressing the Yamanaka factors - and frequently omitting the potent oncogene c-Myc to form an OSK cocktail - researchers can strip away the epigenetic marks of aging, restore youthful gene expression, and repair damaged chromatin architecture without pushing the cell past the threshold of dedifferentiation 818454840.
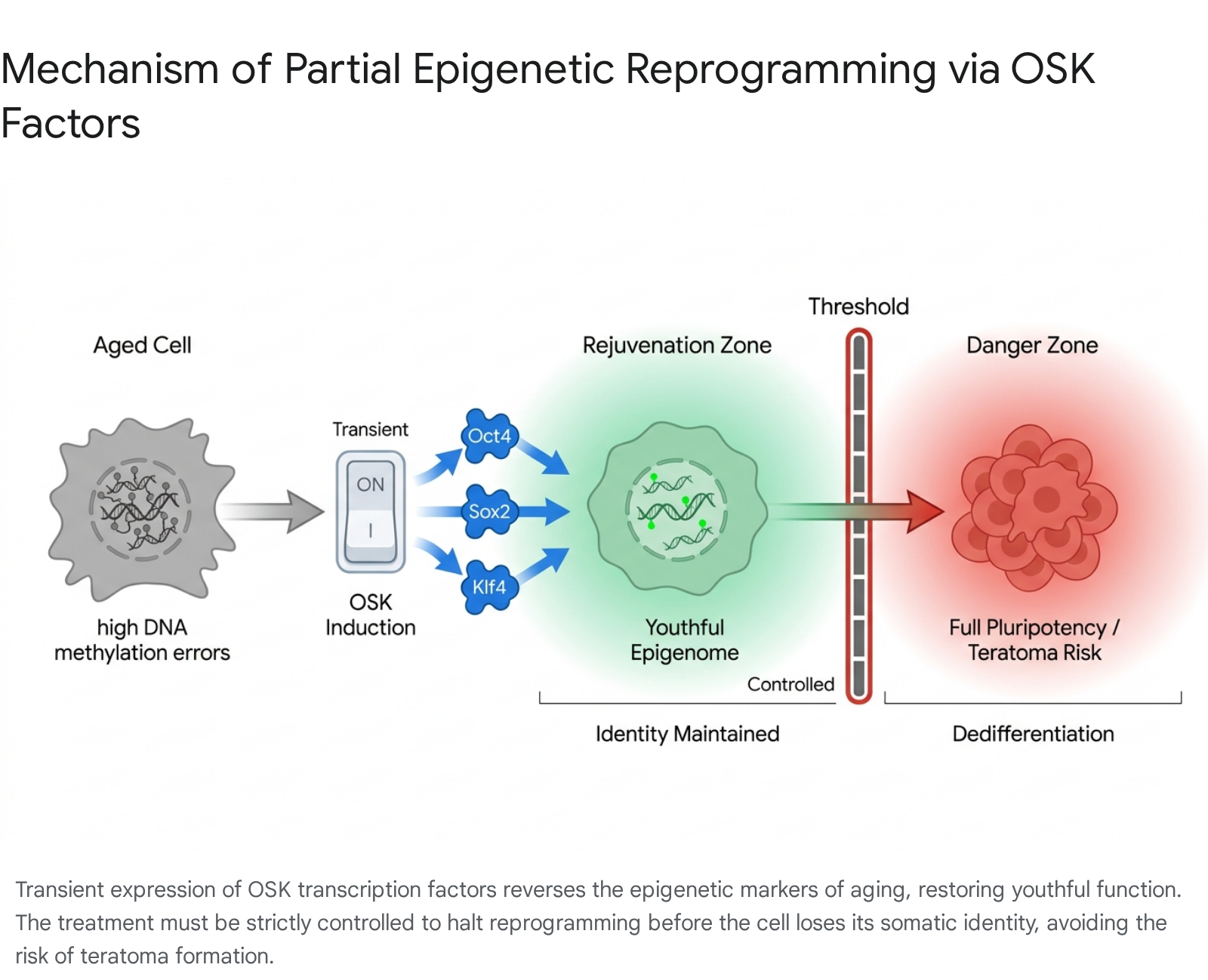
Preclinical Efficacy and Delivery Challenges
Partial reprogramming successfully restores youthful metabolic profiles and regenerative capacities to aged mesenchymal and muscle stem cells 840. It has also demonstrated significant potential in reversing disease states; preclinical studies of partial reprogramming in models of metabolic dysfunction-associated steatohepatitis (MASH) - a severe fibrotic liver disease - showed significant improvements in clinically derived biomarkers and histopathology 41. Remarkably, a recent study demonstrated that systemic adeno-associated virus (AAV)-mediated delivery of OSK factors in exceptionally old wild-type mice extended median remaining lifespan by 109%, marking one of the most profound demonstrations of whole-organism age reversal to date 42.
Translating partial reprogramming from animal models to human therapies involves overcoming significant delivery and safety hurdles. Small-molecule chemical reprogramming cocktails offer an alternative to viral gene therapy, theoretically allowing for simpler oral or topical dosing. However, chemical reprogramming currently suffers from off-target effects and stochastic inefficiency; only a fraction of target cells respond predictably, raising serious dosing concerns 9. Consequently, the local delivery of viral vectors to isolated organs remains the most clinically viable and safe strategy. By targeting specific, enclosed niches, researchers can tightly control the dosage and restrict the spatial impact of the Yamanaka factors, minimizing the systemic risk of tumorigenesis 948.
Clinical Translation and Global Research Initiatives
The transition of niche-restoring senolytics and intrinsic partial reprogramming from bench to bedside is accelerating rapidly, supported by shifting regulatory frameworks and aggressive multinational funding strategies.
FDA Clearances and Ocular Neuropathy Trials
The regulatory landscape for cellular rejuvenation crossed a historic threshold in January 2026, when the U.S. Food and Drug Administration (FDA) cleared an Investigational New Drug (IND) application for Life Biosciences' ER-100 415243. ER-100 is an in vivo gene therapy utilizing an AAV2 vector to deliver OSK factors directly into the eye via intravitreal injection 5244.
The Phase 1 clinical trial is designed to treat serious optic neuropathies, specifically open-angle glaucoma (OAG) and non-arteritic anterior ischemic optic neuropathy (NAION) 414445. By targeting retinal ganglion cells - central nervous system neurons previously thought to be irreversibly lost to aging and damage - ER-100 seeks to directly rejuvenate these cells and restore vision 52. Because the FDA does not formally recognize "aging" as a disease indication, anchoring the reprogramming platform to recognized, localized diseases provided the necessary regulatory pathway to initiate human trials for age reversal technologies 4856. Should ER-100 demonstrate safety and efficacy, the platform is slated for expansion into other indications, potentially utilizing accelerated platform technology approval pathways to target liver diseases 4841.
Accelerated Regulatory Pathways in Japan
Japan, facing immense demographic pressure from an increasingly aged population, has positioned regenerative medicine as a central pillar of its "Society 5.0" national priority initiative 4647. Building on the Nobel Prize-winning discovery of iPSCs at Kyoto University, Japanese regulatory bodies have implemented highly accelerated approval pathways for cellular therapies 46.
In early 2026, Japan's Ministry of Health, Labor, and Welfare endorsed conditional, time-limited approvals for the world's first medical products derived from reprogrammed human cells. This included Amusepri, a therapy developed under the direction of Kyoto University researchers involving the surgical implantation of lab-grown neuronal precursor cells into the brains of Parkinson's disease patients, and ReHeart, an iPSC-derived cardiac cell therapy designed for severe heart failure 46484950. These physician-led trials demonstrate a unique bioethical and regulatory agility, allowing Japanese biotechnology firms to rapidly iterate and commercialize stem cell therapies aimed at circumventing systemic aging decline 4649.
European Consortia and Systemic Rejuvenation
In Europe, long-term multinational research initiatives are systematically unraveling the complexities of stem cell aging. The European Union's Horizon Europe program recently funded the REJUVIMMUNE project (2026 - 2028), coordinated by the Karolinska Institutet in Sweden. This project is dedicated to counteracting the aging of the hematopoietic system. By developing highly localized mRNA therapeutics and partial reprogramming mimicking cocktails (PRMCs), REJUVIMMUNE aims to transiently reprogram HSCs to reverse myeloid skewing and restore youthful immunosurveillance without the risks associated with systemic viral vectors 5152.
Simultaneously, institutions such as the Max Planck Institute for Biology of Ageing are leveraging cross-species genetic mapping and advanced computational modeling to define exactly how the breakdown of specific cellular niches drives systemic decline across organisms 53545556. These European frameworks mirror a growing global consensus: addressing single diseases of aging sequentially is ultimately inefficient; instead, next-generation therapeutics must target the underlying stem cell exhaustion that drives multi-organ failure.
Conclusion
The exhaustion of adult stem cell pools is a foundational mechanism of biological aging, driving a cascade of systemic failures from immunosenescence and sarcopenia to neurodegeneration. This decline is not a unilateral process but a dual-faceted pathology, driven simultaneously by the intrinsic accumulation of molecular damage within the stem cells and the extrinsic corruption of their microenvironmental niches by inflammatory, senescent cells.
The convergence of two distinct therapeutic modalities offers a comprehensive strategy for tissue restoration. Senotherapeutics - ranging from targeted small molecules like dasatinib and navitoclax to engineered CAR T-cells - provide the mechanism to clear the hostile, SASP-laden niche, restoring a physical and biochemical environment conducive to regeneration. Concurrently, partial epigenetic reprogramming via Yamanaka factors offers the unprecedented ability to erase intrinsic epigenetic drift, resetting stem cells to a youthful, functional state without sacrificing their somatic identity. As these technologies move through landmark human clinical trials, they represent a profound shift in medicine: moving from the chronic management of age-related symptoms to the direct biological reversal of the underlying cellular decline.