Sirtuin biology and the epigenetic theory of aging
The Molecular Architecture of Sirtuin Catalysis
The mammalian sirtuin family represents a highly conserved lineage of class III histone deacetylases that has fundamentally reshaped the trajectory of gerontological research over the past three decades. Originally identified in Saccharomyces cerevisiae as the Silent Information Regulator 2 (Sir2) gene - a critical mediator of replicative lifespan via the suppression of extrachromosomal ribosomal DNA circles - sirtuins have since been characterized in all domains of life, including bacteria, archaea, and metazoans 12. In mammals, the family comprises seven distinct paralogs, designated SIRT1 through SIRT7, which exhibit diverse subcellular localizations, distinct enzymatic capacities, and specialized biological functions 34. Sirtuins are unique among deacetylases due to their absolute dependency on nicotinamide adenine dinucleotide (NAD+) as a co-substrate, a biochemical requirement that tightly couples their enzymatic activity to the metabolic and redox states of the cell 55.
The structural architecture of all sirtuins centers around a highly conserved catalytic core domain of approximately 275 amino acids 5. High-resolution crystallographic studies, which currently include over 40 distinct sirtuin structures in various liganded forms, reveal that this core consists of a large Rossmann fold domain - characteristic of NAD+-binding proteins - and a smaller, structurally variable zinc-binding domain 6. The active site is located within the deep cleft between these two domains, forming the precise microenvironment required for the dual binding of the NAD+ co-substrate and the acylated protein target 67.
Kinetic Intermediates and Nicotinamide Inhibition
The catalytic mechanism of sirtuin-mediated deacylation is highly complex. Catalysis initiates with the binding of the acylated substrate (such as an acetylated lysine residue on a histone tail or a transcription factor) and NAD+ 68. Steady-state and rapid-quench kinetic analyses indicate that for SIRT1, SIRT2, and SIRT3, the enzyme must bind the acyl-substrate prior to NAD+ 8. In contrast, SIRT6 is unique among mammalian sirtuins in its capability to bind the acyl-substrate or NAD+ in a random order 8. Conformational shifts, specifically the shifting of the zinc-binding domain toward the Rossmann fold during NAD+ entry, trap the substrates in a productive orientation 79.
The initial chemical step involves the cleavage of the glycosidic bond of NAD+, which is proposed to proceed through an SN1-like mechanism 6. Nucleophilic attack by the acyl oxygen of the acetyl-lysine onto the 1′-carbon of the nicotinamide ribose generates a critical C1′-O-alkylimidate intermediate 8. A conserved histidine residue within the active site acts as a general base to activate the 2′-hydroxyl group of the NAD+ ribose, facilitating the subsequent transfer of the acyl group to the ADP-ribose moiety 8. This strictly coupled reaction yields three products: the deacetylated target protein, nicotinamide (NAM), and a unique metabolite, 2′-O-acetyl-ADP-ribose (OAADPr) 56.
The equimolar consumption of NAD+ for every acyl group removed means that sirtuin activity is highly energy-intensive, directly draining the cellular NAD+ pool during periods of high deacetylation demand 5. Furthermore, the reaction product NAM acts as a potent noncompetitive inhibitor of sirtuins 610. NAM binds to a specific "C-pocket" within the sirtuin catalytic cleft, lying adjacent to the NAD+-binding pocket, and promotes a reverse base-exchange reaction that aborts the deacetylation process 1011. Susceptibility to NAM inhibition is heavily dependent on the specific acyl-substrate chain length; for instance, SIRT6 displays its greatest susceptibility to NAM inhibition in the presence of myristoylated and decanoylated peptides relative to hexanoylated substrates 8. To maintain sirtuin activity, cells must continually clear NAM via the NAD+ salvage pathway, utilizing the rate-limiting enzyme nicotinamide phosphoribosyltransferase (NAMPT) to recycle NAM back into nicotinamide mononucleotide (NMN), and ultimately NAD+ 11.
Mammalian Sirtuin Isoforms and Functional Specialization
Mammalian sirtuins have evolved distinct N-terminal and C-terminal extensions that govern their subcellular targeting, enzymatic specificities, and protein-protein interactions. The evolutionary divergence of these paralogs allows them to partition metabolic and epigenetic regulation across different cellular compartments, adapting the organism to various forms of metabolic, oxidative, and genotoxic stress 14.
| Sirtuin Isoform | Subcellular Localization | Primary Enzymatic Activities | Key Physiological and Longevity Functions |
|---|---|---|---|
| SIRT1 | Nucleus, Cytosol | Strong Deacetylation | Regulation of metabolic sensors (AMPK, PGC-1α), DNA repair coordination, apoptosis regulation (p53, FOXO), and circadian rhythm maintenance. |
| SIRT2 | Cytosol, Nucleus | Strong Deacetylation, Defatty-acylation | Cell cycle control, microtubule dynamics, and master regulation of aging-associated inflammation (NF-κB, NLRP3 inflammasome, cGAS-STING). |
| SIRT3 | Mitochondria (Matrix) | Strong Deacetylation | Mitochondrial homeostasis, oxidative phosphorylation regulation, ROS mitigation via SOD2, and prevention of ferroptosis. |
| SIRT4 | Mitochondria | ADP-ribosylation, Lipoamidase | Inhibition of glutamine catabolism and fatty acid oxidation; acts as a negative regulator of cardiomyocyte proliferation. |
| SIRT5 | Mitochondria | Desuccinylation, Demalonylation | Urea cycle regulation, maintenance of fatty acid catabolism via ECHA, and preservation of cardiac function. |
| SIRT6 | Nucleus | Deacetylation, ADP-ribosylation | DNA double-strand break repair, telomere maintenance, suppression of LINE-1 retrotransposons; recognized as the primary longevity sirtuin. |
| SIRT7 | Nucleus (Nucleolus) | Deacetylation, Desuccinylation | Ribosome biogenesis, rRNA transcription, DNA repair via NHEJ; exhibits paradoxical context-dependent lifespan extension in male mice. |
Sirtuin 1 and the Regulation of Metabolic Homeostasis
SIRT1, the most extensively studied mammalian ortholog of yeast Sir2, acts primarily as a nucleocytoplasmic energy sensor that coordinates metabolic shifts and restrains pro-inflammatory signaling 513. Transgenic mice globally overexpressing SIRT1 generally exhibit robust protection against diet-induced metabolic syndrome, hepatic steatosis, and certain malignancies; however, unlike other models, they do not typically show significant increases in maximum absolute longevity under standard dietary conditions 514. SIRT1's primary mechanism involves the deacetylation and subsequent activation of critical transcription factors and co-activators. It deacetylates PGC-1α to drive mitochondrial biogenesis, modulates the FOXO family (FOXO1, FOXO3a, FOXO4) to enhance stress resistance, and inhibits the p65 subunit of NF-κB to restrain age-related chronic inflammation, often termed "inflammaging" 41213.
Additionally, SIRT1 serves a crucial bridging function in the DNA damage response (DDR) by deacetylating DNA repair proteins such as Ku70 and NBS1, thereby recruiting them to DNA damage foci 1415. SIRT1 also exhibits complex, tissue-specific crosstalk with AMP-activated protein kinase (AMPK). SIRT1 can activate AMPK by deacetylating its upstream kinase, LKB1, while AMPK reciprocally activates SIRT1 by increasing the intracellular NAD+/NADH ratio, establishing a positive feedback loop crucial for energy homeostasis 1113.
Sirtuin 2 and the Control of Inflammaging
SIRT2 is primarily cytosolic but dynamically translocates to the nucleus during specific phases of the cell cycle 35. Recent gerontological research has positioned SIRT2 as a master regulator of aging-associated inflammation. Aged SIRT2 knockout mice exhibit severely compromised muscle function, cognitive decline, stem cell exhaustion, and marked susceptibility to severe viral infections like COVID-19 16. SIRT2 mitigates inflammation by directly deacetylating key components of multiple immune pathways, including the NLRP3 inflammasome, the cGAS-STING signaling axis, and STAT3 16. Pharmacological interventions that boost systemic NAD+ levels, such as the CD38 inhibitor 78c, have been shown to powerfully suppress aging-associated inflammation and improve tissue function in aged wild-type mice, an effect largely mediated by the restoration of SIRT2 catalytic activity 16.
Mitochondrial Dynamics: Sirtuins 3, 4, and 5
The mitochondrial matrix harbors SIRT3, SIRT4, and SIRT5, which collectively orchestrate the organelle's metabolic output and defensive responses 15. SIRT3 acts as the primary mitochondrial deacetylase. By deacetylating metabolic enzymes, SIRT3 optimizes the electron transport chain, reduces reactive oxygen species (ROS) leakage, and drives the mitochondrial unfolded protein response 2017. It provides a critical defensive repertoire against ferroptosis - a catastrophic failure of cellular lipid quality control - by preserving mitochondrial membrane integrity and synergistically activating the antioxidant enzyme superoxide dismutase 2 (MnSOD/SOD2) 17.
Interestingly, SIRT4 has emerged as a negative regulator of specific longevity pathways. Unlike SIRT3, SIRT4 possesses negligible deacetylase activity in vitro, functioning predominantly as an ADP-ribosyltransferase and lipoamidase 518. SIRT4 inhibits glutamine catabolism by ADP-ribosylating glutamate dehydrogenase (GDH) and suppresses fatty acid oxidation by downregulating malonyl-CoA decarboxylase and PPARα 1819. During caloric restriction, when SIRT3 and SIRT5 are upregulated to promote survival and fatty acid oxidation, SIRT4 is actively downregulated 1820. Kinetic studies reveal that SIRT4 is the primary mammalian sirtuin physiologically regulated by the absolute NAD+/NADH ratio rather than by NAD+ concentration alone; it is inhibited by physiological concentrations of NADH 20. Recent studies utilizing cardiac models reveal that SIRT4 expression increases with age and that its overexpression suppresses cardiomyocyte proliferation by inducing oxidative DNA damage 21. Remarkably, SIRT4 deficiency promotes cardiomyocyte proliferation and extends the regenerative window of the neonatal heart, establishing it as an antagonistic factor in post-ischemic tissue repair 21.
Sirtuin 6: The Primary Mammalian Longevity Regulator
Among the mammalian paralogs, SIRT6 is currently the most unequivocally validated longevity regulator. Complete targeted deletion of SIRT6 in mice results in a severe, highly penetrant progeroid (premature aging) phenotype, characterized by genomic instability, massive metabolic defects, severe hypoglycemia, and early death usually within weeks of birth 1922. Conversely, whole-body overexpression of SIRT6 significantly extends lifespan in male mice by approximately 14.5%, making it the sole sirtuin whose isolated genetic overexpression robustly extends mammalian lifespan 1427.
SIRT6 functions dually as a deacetylase and a mono-ADP-ribosyltransferase, acting as the ultimate gatekeeper of chromatin integrity 23. It continuously anchors to chromatin and is rapidly mobilized to DNA double-strand breaks (DSBs) 23. This mobilization is facilitated synergistically by SIRT1, which deacetylates SIRT6 at residue K33; this deacetylation promotes SIRT6 polymerization and its recruitment to γH2AX foci at sites of damage 2324. Once localized, SIRT6 recruits essential repair factors such as Ku80, the 9-1-1 complex, and PARP1 23. Crucially, SIRT6 transiently suppresses local transcription at the damage site by mono-ADP-ribosylating the histone demethylase KDM2A, leading to its displacement from chromatin 25. This localized transcriptional pausing ensures that RNA polymerase II does not collide with the non-homologous end-joining (NHEJ) repair machinery, thereby preventing catastrophic genomic instability and carcinogenic translocations 25.
The Paradoxical Lifespan Effects of Sirtuin 7
The nucleolar sirtuin, SIRT7, dictates RNA polymerase I activity and governs ribosome biogenesis 526. Early studies indicated that SIRT7-deficient mice suffered from partial embryonic lethality and a progeroid-like phenotype characterized by impaired repair of DSBs and genomic instability 2728. However, rigorous independent re-evaluations published in late 2025 present a conflicting and highly paradoxical narrative: male SIRT7 knockout mice on a pure C57BL/6J background actually demonstrated a significant extension in both mean and maximum lifespan, alongside a marked delay in age-associated mortality 2930.
This lifespan extension appears to be driven by an unexpected systemic metabolic shift. The knockout mice exhibited greatly elevated serum levels of Fibroblast Growth Factor 21 (FGF21), an endocrine hormone known to enhance insulin sensitivity and promote metabolic flexibility 29. Activating transcription factor 4 (ATF4) stimulates FGF21 transcription, and hepatic levels of ATF4 mRNA were found to be massively upregulated in aged male SIRT7 knockout mice compared to wild-type controls 29. The paradox of SIRT7 - acting as an essential mediator of DNA repair and a potential oncogene in specific advanced cancers, while its global suppression extends male longevity via systemic metabolic remodeling - highlights the extreme complexity of sirtuin biology 282930. It is increasingly evident that not all class III deacetylases act as unidirectional anti-aging effectors, and their roles are heavily dependent on tissue context, age, and biological sex 30.
The Rise, Fall, and Rehabilitation of Sirtuin-Activating Compounds
The hypothesis that sirtuins could be pharmacologically activated to mimic the profound life-extending effects of caloric restriction led to the aggressive, multi-billion-dollar pursuit of Sirtuin-Activating Compounds (STACs). The discovery, subsequent backlash, and ultimate structural rehabilitation of these molecules underscore the immense complexities of translating in vitro enzymology into clinical therapeutics.
Initial Discoveries and the Artifact Hypothesis
In the early 2000s, high-throughput screens utilizing recombinant human SIRT1 identified resveratrol - a natural polyphenol found in grapes and red wine - as a potent allosteric activator 35. Resveratrol was observed to increase the catalytic efficiency of SIRT1 by up to eightfold, functioning by significantly lowering the Michaelis constant (Km) for its target peptide substrates without altering the maximum reaction velocity (Vmax) 511. This discovery rapidly catalyzed the development of more potent, bioavailable synthetic STACs, including imidazothiazoles (such as SRT1720), thiazolopyridines (STAC-2), benzimidazoles (STAC-5), and bridged ureas (STAC-9) 10. In preclinical models, these synthetic compounds exhibited massive therapeutic promise, effectively mitigating diet-induced obesity, improving insulin sensitivity, and extending the lifespan of mice fed high-calorie diets 510.
However, the field soon encountered a severe methodological crisis that threatened the core premise of the longevity theory. Independent biochemists demonstrated that the initial high-throughput screening assays utilized a synthetic peptide substrate artificially tagged with a bulky, hydrophobic fluorophore - 7-amino-4-methylcoumarin (AMC) - to optically measure deacetylation rates 931. When these critics tested resveratrol and early synthetic STACs against native, untagged peptide sequences, the compounds completely failed to activate SIRT1 9. This discrepancy birthed the "artifact hypothesis," which posited that STACs were merely binding to the artificial fluorophore rather than structurally agonizing the enzyme, rendering them biologically irrelevant artifacts of the assay design 2731. The ensuing controversy deeply fractured the aging research community and contributed to the termination of several high-profile pharmaceutical development programs, including GlaxoSmithKline's suspension of clinical trials for early STAC candidates 2737.
Structural Resolution of the Allosteric Binding Mechanism
The resolution of the STAC controversy required years of advanced structural biology and kinetic analyses. Researchers eventually demonstrated that while STACs indeed required the AMC fluorophore to activate SIRT1 against extremely short, synthetic peptides, this was solely because the bulky fluorophore mimicked the presence of native hydrophobic amino acid residues typically found adjacent to acetylated lysines in authentic, full-length biological substrates 9. When researchers substituted the artificial fluorophore with the large, hydrophobic amino acids naturally present in endogenous SIRT1 targets - such as PGC-1α and FOXO3a - resveratrol and synthetic STACs successfully activated the enzyme in vitro 9.
X-ray crystallography ultimately provided incontrovertible proof that STACs operate via a true allosteric mechanism 1131. SIRT1 contains a unique N-terminal STAC-binding domain (SBD) extending from residues 183 to 229, an unstructured region completely absent in other mammalian sirtuins 911. Crystallographic data of a truncated SIRT1 bound to STAC-1 and an AMC-containing peptide revealed that STAC molecules bind precisely to the SBD. They act as a physical structural bridge that stabilizes the interaction between the enzyme's N-terminal domain and the central catalytic core 1131. This allosteric bridge closes the active site cleft, markedly increasing the enzyme's binding affinity for specific substrates bearing hydrophobic motifs 1131. Further genetic validation was achieved by identifying a single-amino-acid substitution in the SIRT1 catalytic core (Glu230 to Lys) that completely abolished activation by resveratrol and over 100 structurally diverse synthetic STACs, confirming a common, specific allosteric binding pocket 1011.
Next-Generation Sirtuin Activators
While SIRT1 activators dominated early research, the evolutionary divergence of sirtuin active sites meant that drugs designed for SIRT1 largely failed to activate other isoforms 37. Recent pharmacology has pivoted toward targeting alternative sirtuins, particularly those in the mitochondria. Because SIRT3 lacks an identified N-terminal SBD homologous to SIRT1, it was long considered "undruggable" through traditional allosteric activation 3738.
However, in late 2024 and 2025, researchers at Chakrabarti Capital Management (CCM) Biosciences utilized advanced computational screening to identify novel SIRT3 activators (e.g., compounds 5329973 and 5689785) 38. These molecules are capable of nearly doubling SIRT3 activity against endogenous targets like MnSOD. Critically, these novel compounds fully recover SIRT3 catalytic velocity even when ambient mitochondrial NAD+ levels are reduced by half, simulating the severe NAD+ depletion characteristic of aging tissue 3738. These compounds have reportedly outperformed existing NAD+ supplements in preclinical murine models and are slated for clinical trials targeting Alzheimer's disease and age-related infertility in late 2025 and 2026 3738.
The Information Theory of Aging and the ICE Model
The biological functions of sirtuins, particularly their dual roles in regulating epigenetic gene expression and actively relocating to repair DNA damage, directly informed Dr. David Sinclair's comprehensive "Information Theory of Aging" (ITOA) 3233. The theory posits a fundamental paradigm shift: aging is not primarily driven by the irreversible accumulation of genetic mutations (the Somatic Mutation Theory), but rather by the progressive, systemic degradation of analog epigenetic information 324134.
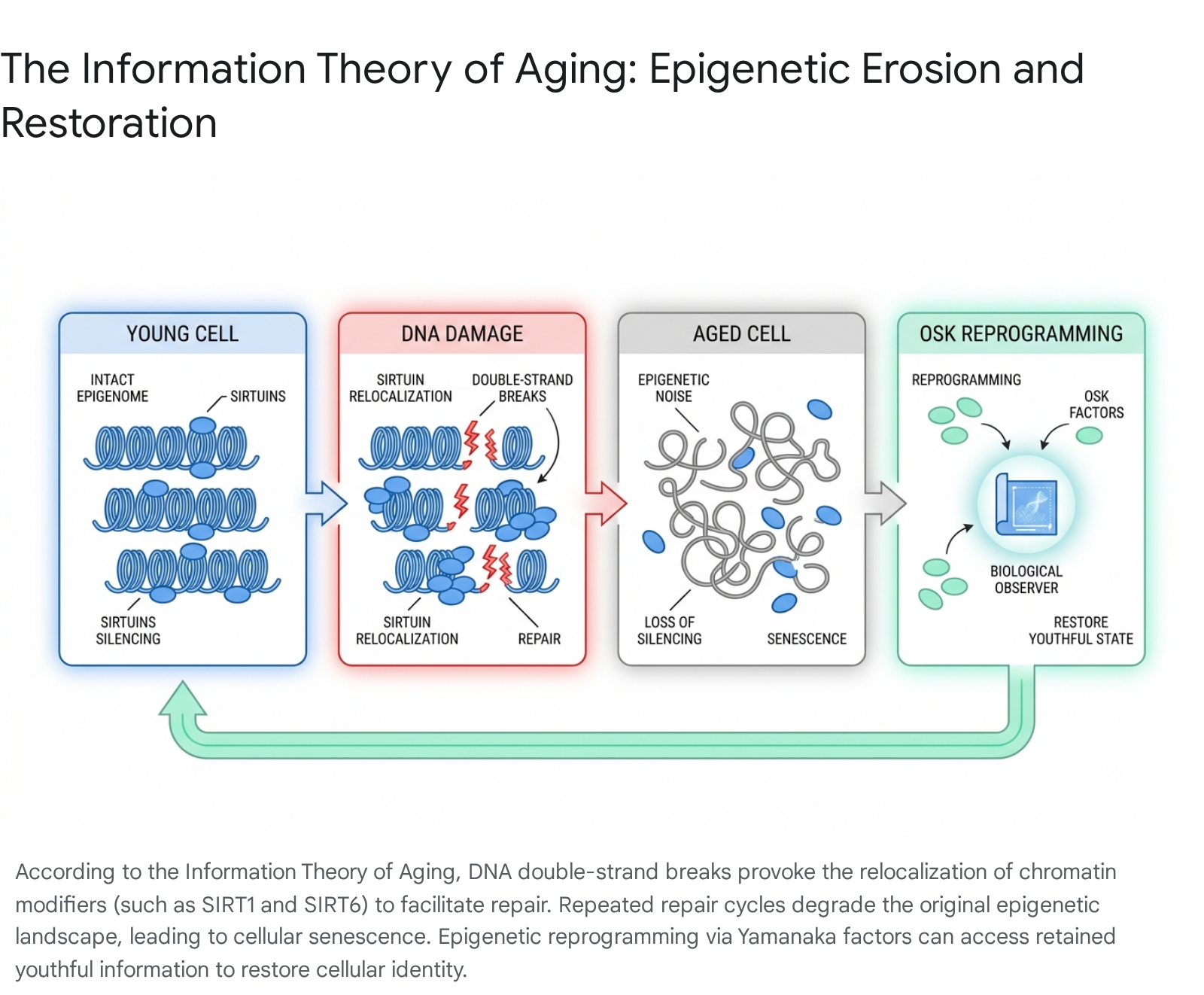
Epigenetic Noise and the Relocalization Hypothesis
According to the ITOA, cells suffer continuous DNA damage - specifically double-strand breaks (DSBs) - from environmental exposure, oxidative stress, and endogenous metabolism 152343. Repairing this severe damage requires chromatin-modifying proteins, prominently including SIRT1 and SIRT6, to physically leave their normal genomic binding sites (where they suppress transcription of specific loci and maintain heterochromatin) and relocate to the damage sites to facilitate the DNA damage response 152343. While the DNA sequence is effectively repaired (maintaining high genetic fidelity), the chromatin modifiers do not always return to their exact original positions 2344.
Over decades of life, this repeated cycle of damage and relocalization generates profound "epigenetic noise." The precise cellular epigenetic landscape, initially established during embryogenesis, begins to flatten; normally silenced genomic regions are inappropriately transcribed, and essential active genes are suppressed 2345. Consequently, cells gradually lose their distinct functional identities, a phenomenon termed cellular exdifferentiation, and eventually enter an irreversible state of senescence 4435.
The Inducible Changes to the Epigenome Mouse Model
To empirically validate the ITOA, Sinclair's laboratory engineered the "ICE" (Inducible Changes to the Epigenome) mouse model 4435. These mice carry a tamoxifen-inducible transgene expressing the I-PpoI endonuclease, an enzyme that generates highly specific, non-mutagenic DSBs at exactly 19 non-coding regions and one specific ribosomal DNA (rDNA) sequence in the mammalian genome 363749. Because I-PpoI creates "sticky" 4-basepair overhangs, the resulting breaks have extremely low mutagenic potential and are repaired seamlessly 37. By inducing these precise breaks, the researchers experimentally accelerated the requirement for DNA repair without increasing the burden of actual genetic mutations, effectively isolating epigenetic disruption from genetic damage 4337.
The phenotypic results, published comprehensively in the journal Cell in 2023, were highly striking. Following the induction of the endonuclease and subsequent repair cycles, ICE mice experienced a severe acceleration of physiological aging. After ten months, the treated cohort demonstrated hair loss, reduced body mass, cognitive decline, muscle degeneration, and severe visual impairment 4137. System-specific age-mimicking phenotypes were observed at the level of kidney architecture and increased glial cell activation in the brain 37. Molecularly, their DNA methylation patterns dramatically accelerated, shifting their epigenetic clocks to match those of chronologically much older wild-type mice 4144. The induction of this progeroid phenotype, driven purely by mutation-less DSBs, provided robust evidence that the disruption of the epigenome alone is sufficient to drive mammalian aging 4337.
Epigenetic Reprogramming and the Biological Observer
If aging fundamentally constitutes analog epigenetic corruption, the ITOA posits that it must be reversible. Drawing upon Claude Shannon's mathematical theory of communication, Sinclair theorized the existence of a cellular "biological observer" - a safely sequestered, retained backup copy of youthful epigenetic information established during embryogenesis 334350.
To test whether this backup information could be accessed, the researchers utilized adeno-associated viruses (AAVs) to ectopically express three of the four Yamanaka reprogramming transcription factors (Oct4, Sox2, and Klf4, collectively referred to as "OSK") in the aged and damaged ICE mice 334551. Crucially, the oncogenic c-Myc factor was excluded to prevent tumorigenesis 33. Transient OSK expression successfully reset the epigenetic age of the cells, re-silenced dysregulated genes, and restored physiological function. In murine models of natural aging and induced nerve crush injuries, OSK therapy reversed age-related vision loss and successfully regenerated severed retinal ganglion cell (RGC) axons 3351. Importantly, this epigenetic rejuvenation required the active functions of the DNA demethylases TET1 and TET2, confirming that active rewriting of the DNA methylome is absolutely essential for retrieving the "observer" data and restoring cellular identity 45. The exact structural or molecular nature of this observer mechanism - whether it relies on DNA-RNA hybrids, long-lived chromatin complexes, or specific combinations of histone marks - remains the subject of intense ongoing investigation 3343.
Competing Theoretical Frameworks and Peer Critique
The publication of the ICE model and the formalization of the ITOA provoked significant debate and critical appraisal within the gerontology community 3839. Peer-reviewed critiques published in late 2024 and 2025 challenged whether the ITOA fully accounts for the vast heterogeneity of the aging process.
Proponents of Mikhail Blagosklonny's "Hyperfunction Theory" argue that aging is not merely the passive accumulation of molecular damage or epigenetic noise, but rather a quasi-programmed, maladaptive continuation of developmental growth pathways 5040. Under this model, the chronic, persistent overactivation of nutrient-sensing pathways like mTOR - driving cells toward hypertrophy and senescence long after developmental maturity is reached - is the primary driver of age-related pathology 40. Consequently, these critics suggest the ITOA's focus on DNA damage as the initiator of aging is overly narrow 40.
Furthermore, critics have pointed out that the I-PpoI endonuclease model, while elegantly demonstrating the consequences of massive, simultaneous DSB repair, represents an acute supraphysiological stress that may not perfectly mirror the slow, stochastic, and endogenous decay characteristic of natural chronological aging 3637. Nonetheless, Sinclair's laboratory defended the model in peer-reviewed responses in Cell, emphasizing that the bidirectional control of the epigenome - both accelerating it forward into senescence through controlled DNA damage and driving it backward into a youthful state via OSK partial reprogramming - provides the strongest evidence to date that the epigenome serves as the primary, modifiable substrate of biological aging 4339.
Clinical Translation: NAD+ Precursor Trials and Meta-Analyses
While in vivo epigenetic reprogramming via viral gene therapy remains years away from safe, approved human application, the immediate clinical translation of sirtuin biology relies heavily on modulating the NAD+ salvage pathway. By supplying exogenous NAD+ precursors, clinicians aim to restore the optimal NAD+/NADH ratio in older adults, thereby reactivating the endogenous network of sirtuin enzymes that have been starved of their required co-substrate 527.
Nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR) have dominated the global nutraceutical market as the premier oral NAD+ precursors 55. However, the human clinical data mapping their efficacy remain highly nuanced and occasionally contradictory. A series of rigorous meta-analyses published in early 2026 synthesize the outcomes of recent randomized controlled trials (RCTs).
A comprehensive 2026 meta-analysis conducted by Zheng et al. and Wang et al. examined trials largely conducted in Asian demographics, assessing the physiological impact of NMN at doses ranging from 250 to 1250 mg/day over durations of 4 to 24 weeks 565741. The total combined sample size for the NMN trials was relatively small, totaling 412 participants 56.
| Clinical Endpoint | 2026 Meta-Analysis Outcome | Efficacy Context and Critical Variables |
|---|---|---|
| Gait Speed & Endurance | Improved | Significant increase observed (approx. +1 ft/s). Improvements were most pronounced in adults aged 40 - 60 years 5659. |
| Hepatic Function (ALT levels) | Improved | Significant reduction in alanine aminotransferase (ALT) levels (up to a 15% decrease), indicating reduced hepatic stress and injury 5659. |
| Insulin Sensitivity (HOMA-IR) | Minimal / Dose-Dependent | Broadly nonsignificant across all ages. However, sub-analyses suggest potential efficacy strictly limited to lower doses (≤ 300 mg/day) 5641. |
| Fasting Glucose & HbA1c | No Significant Effect | No consistent, statistically significant reduction in primary glycemic control markers across the major studied cohorts 41. |
| Grip Strength | Minimal | No broad improvement was observed across the general population; isolated benefits were noted exclusively in participants over 60 years old 56. |
The synthesized data indicate that while NMN is a highly promising therapeutic for specific age-related functional declines - such as skeletal muscle endurance, physical fatigue, and hepatic stress - it does not appear to act as a universal panacea capable of reversing severe, established metabolic syndrome in human cohorts 5641.
Furthermore, researchers highlight that the current evidence base for NAD+ precursors is structurally asymmetric. NMN trials have predominantly featured East Asian cohorts at comparatively lower doses (250 - 300 mg), while NR trials have centered on Western populations at significantly higher doses (up to 2,000 mg daily) 5557. This structural heterogeneity, combined with incompatible assay matrices used to measure whole-blood NAD+ pharmacodynamics across different studies, currently precludes any reliable head-to-head indirect comparisons between NMN and NR efficacy 5557.
The Regulatory Landscape and Combinatorial Interventions
The clinical and commercial trajectory of NMN in the United States was severely disrupted in November 2022. The U.S. Food and Drug Administration (FDA) abruptly declared that NMN was excluded from the definition of a dietary supplement under the Dietary Supplement Health and Education Act (DSHEA) 6061. This controversial decision hinged strictly on the DSHEA "drug preclusion clause." The clause was triggered because the pharmaceutical company MetroBiotech had previously initiated an Investigational New Drug (IND) application and conducted substantial clinical investigations for a proprietary NMN formulation (MIB-626) 60.
Following years of intense industry pushback, aggressive litigation by the Natural Products Association (NPA), and the filing of formal Citizen Petitions, the FDA abruptly reversed its stance in late 2025 and early 2026 6263. In formal letters issued to major ingredient suppliers, the agency acknowledged substantial evidence proving that NMN was lawfully marketed as a dietary supplement prior to the authorization of the new drug investigations (adhering to the "race-to-market" interpretation of the statute) 6061. Consequently, NMN's status as a legal dietary ingredient in the U.S. was fully restored, eliminating a massive regulatory bottleneck and re-opening the consumer market 6162. However, global regulations remain heavily fragmented; the European Union continues to mandate strict "Novel Food" authorization protocols for NMN distribution as of 2026, creating a stark regulatory divide between markets 62.
The Shift Toward Precision and Combinatorial Regimens
As the field of gerontology matures, the simplistic "one-size-fits-all" approach to sirtuin activation is rapidly shifting toward precision pharmacology and combinatorial dietary regimens 20. Emerging research indicates that interventions like vigorous physical exercise and metabolic uncoupling rely heavily on baseline mitochondrial integrity. Consequently, they often fail to yield benefits in advanced-age individuals whose mitochondria are already severely degraded by the aging process 20. In these older cohorts, direct NAD+ boosters are theorized to bypass the initial mitochondrial barrier, re-establishing SIRT3 and SIRT1 activity directly to rebuild baseline energetic capacity 20.
Simultaneously, alternative dietary regimens designed to suppress aging-associated inflammation are undergoing rigorous evaluation. Clinical case series published in 2024 demonstrated that combining alternative, established NAD+ precursors like prolonged-release niacin with AMPK-activators (dihydroberberine) and anti-inflammatory flavonoids (taxifolin) effectively downregulates systemic inflammation and corrects dyslipidemia in octogenarian patients 64. This combinatorial approach resulted in an average 35% decrease in LDL cholesterol and a 52% increase in HDL cholesterol, significantly reducing patient reliance on traditional statin therapies 64.
Conclusion
The biology of sirtuins has evolved dramatically from the initial discovery of yeast Sir2 to the realization that these NAD+-dependent deacylases function as the master architects of mammalian epigenetic and metabolic stability. The early era of sirtuin research was undoubtedly marred by methodological controversy surrounding the specific mechanism of resveratrol and synthetic STACs. However, rigorous structural biochemistry and crystallography ultimately confirmed that specific sirtuins, particularly SIRT1, possess distinct allosteric binding domains capable of potent pharmacological modulation, successfully validating decades of academic pursuit.
Today, the scientific consensus recognizes SIRT6 as the predominant driver of genome stability and longevity, owing to its dual ability to repair DNA double-strand breaks and preserve the epigenome, while mitochondrial sirtuins like SIRT3 defend against metabolic decay and ferroptosis. Sinclair's Information Theory of Aging elegantly integrates these diverse functions, proposing that the ultimate cause of aging is the epigenetic noise generated when chromatin modifiers like sirtuins perpetually relocalize to handle persistent DNA damage. The success of partial OSK reprogramming in reversing this epigenetic noise in the ICE mouse model points toward a future where aging is treated not as the inevitable accumulation of structural damage, but as a potentially curable form of analog data loss. While clinical epigenetic reprogramming advances cautiously through preclinical phases, the optimization of NAD+ precursors, the resolution of regulatory blockades, and the advent of next-generation, isoform-specific allosteric activators represent the most viable, immediate interventions to safely extend human healthspan.