Senescence-associated secretory phenotype
Cellular Senescence and the Secretory Profile
Cellular senescence is a fundamental biological state characterized by an essentially irreversible arrest of the cell cycle, coupled with profound morphological, epigenetic, and metabolic alterations 123. Initially identified as a mechanism limiting the proliferative capacity of human diploid fibroblasts in culture - a phenomenon known as replicative exhaustion or the Hayflick limit - senescence is now understood to be a highly complex, dynamic stress-response program 456. Cells can enter this state in response to a vast array of endogenous and exogenous stressors, including telomere attrition, DNA double-strand breaks, oncogene activation, mitochondrial dysfunction, and severe oxidative stress 789.
While senescent cells definitively lose their capacity to divide, they remain highly metabolically active 1011. Unlike apoptotic cells, which are systematically dismantled and cleared without eliciting a persistent inflammatory response, senescent cells survive indefinitely by upregulating powerful anti-apoptotic pathways, notably those involving the BCL-2 protein family 11213. Concurrently, they undergo a dramatic shift in their intracellular secretory machinery, developing what is formally termed the Senescence-Associated Secretory Phenotype (SASP) 31415.
The SASP represents the primary non-cell-autonomous mechanism through which senescent cells influence their local and systemic microenvironment 14. It is an extensive and highly heterogenous secretome comprising hundreds of soluble and insoluble factors, including pro-inflammatory cytokines, chemokines, growth factors, matrix-remodeling proteases, bioactive lipids, and extracellular vesicles 14161718. Rather than acting as inert, non-dividing entities in tissue, senescent cells utilize the SASP to communicate aggressively with neighboring parenchymal, stromal, and immune cells 19.
The evolutionary logic underlying the SASP appears to be inherently dualistic, reflecting the principles of antagonistic pleiotropy 71220.
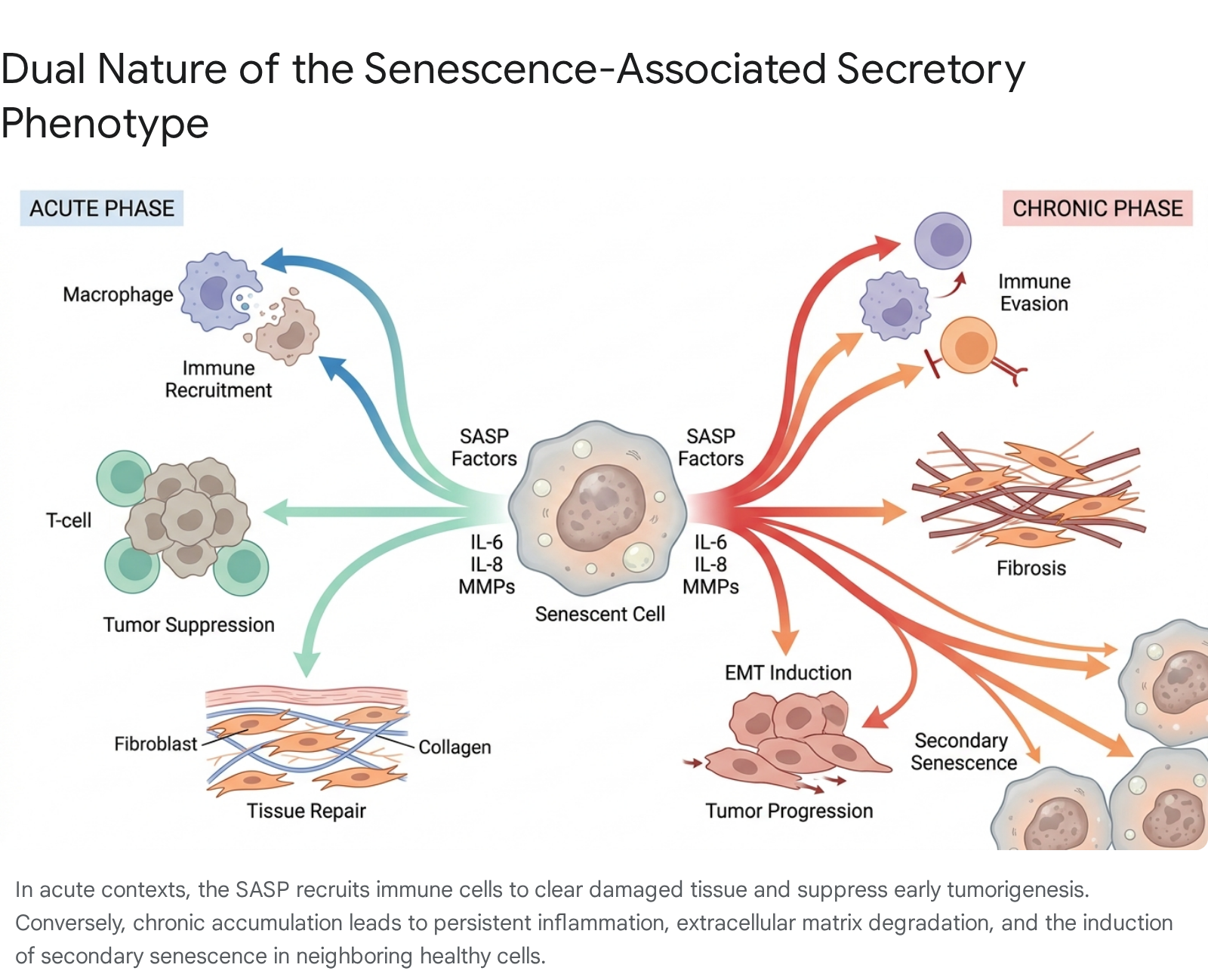
In transient, acute physiological contexts - such as embryonic development or immediate tissue injury - the SASP acts as a critical beacon, recruiting cells of the innate and adaptive immune systems to clear damaged cells and orchestrating tissue regeneration through the stimulation of local progenitor cells 2715. However, when senescent cells resist clearance and accumulate chronically in tissues, as is characteristic of advancing biological age, the persistent secretion of SASP factors establishes a maladaptive, low-grade inflammatory microenvironment 1215. This prolonged state of "inflammaging" actively drives tissue destruction, functional decline, and the pathogenesis of a myriad of age-related conditions, including neurodegeneration, cardiovascular disease, osteoarthritis, and cancer 3151921.
Biological Mechanisms of Senescence Arrest
The establishment of the senescent state and the subsequent induction of the SASP are tightly regulated by overlapping tumor suppressor networks and DNA damage response (DDR) pathways 18. The core mediators of cell cycle arrest are the p53/p21 (CDKN1A) and the p16INK4a (CDKN2A)/retinoblastoma (Rb) pathways 2816. Upon detection of critical telomere shortening or persistent DNA damage, ataxia-telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) kinases are recruited, initiating a phosphorylation cascade involving CHK1 and CHK2 that stabilizes the p53 transcription factor 91618. Elevated p53 upregulates the cyclin-dependent kinase inhibitor p21, which halts cell cycle progression and prevents DNA replication 8.
Simultaneously or sequentially, the p16INK4a pathway acts to inhibit cyclin-dependent kinases 4 and 6 (CDK4/6), preventing the phosphorylation of the Rb protein 922. Hypophosphorylated Rb actively represses E2F transcription factors, reinforcing the permanent block on the cell cycle 28. While these pathways secure the proliferative arrest, they operate somewhat independently from the mechanisms that generate the SASP. For instance, severe genotoxic stress triggers both arrest and the SASP, but certain developmental senescent programs execute cell cycle arrest via p21 without widespread DNA damage, leading to a modified, transient secretory profile without systemic inflammation 2.
Mitochondrial dysfunction has emerged as a distinct, unappreciated hallmark that cements the senescent phenotype 715. Damaged mitochondria in senescent cells release reactive oxygen species (ROS) and cytosolic chromatin fragments 71518. These fragments are detected by the cGAS-STING innate immune pathway, which interprets the cytosolic DNA as a pathogen-like signal. The sustained activation of cGAS-STING further hyper-activates downstream inflammatory cascades, intimately linking mitochondrial breakdown with the maintenance of the chronic SASP, even after primary DNA damage signaling has plateaued 815.
Molecular Composition of the Secretory Phenotype
The SASP is not a single, static molecular signature but rather a highly dynamic network of secreted factors 31014. Proteomic and transcriptomic profiling of senescent cells reveals significant heterogeneity depending on the tissue of origin, the specific senescence-inducing stressor, and the temporal stage of senescence 101417. However, several core components remain remarkably conserved across most senescent cell lineages.
Interleukins and Inflammatory Cytokines
Pro-inflammatory cytokines constitute the most robust and widely recognized features of the SASP 1017. Interleukin-6 (IL-6) and interleukin-8 (IL-8, also known as CXCL8) are frequently cited as the primary effectors of senescence-associated immune activation and microenvironmental signaling 14. IL-6 operates through both autocrine pathways - binding to the cell's own receptors to reinforce the senescent growth arrest - and paracrine pathways, driving systemic chronic inflammation and inhibiting normal cellular differentiation 81923.
Interleukin-1 alpha (IL-1α) and Interleukin-1 beta (IL-1β) play critical roles as upstream master regulators of the SASP 1417. IL-1α, in particular, often remains tethered to the plasma membrane and acts as a primary signal that activates the Nuclear Factor kappa B (NF-κB) transcription factor 2324. This triggers a massive amplification loop of downstream cytokines, generating the hypersecretory state characteristic of mature senescence 2324. Prolonged exposure to these cytokines in aging tissues impairs local stem cell function, induces insulin resistance in adipocytes, and disrupts overarching structural tissue homeostasis 1025.
Chemokines and Immune Modulators
Chemokines are secreted heavily by senescent cells to direct the migration and localization of immune cells 1417. The CXC motif ligand family (including CXCL1, CXCL2, CXCL3, and CXCL10) and CC motif ligands (such as CCL2, also known as Macrophage Chemoattractant Protein-1 or MCP-1) are prominent components 171826.
In a healthy physiological setting, the secretion of CCL2 and CXCL1 is vital for recruiting natural killer (NK) cells, monocytes, and macrophages to the site of damage, facilitating the targeted phagocytosis of the senescent cell itself - a process termed immune surveillance 1018. However, in a pathological setting where senescent cells evade clearance and accumulate, the continuous hyper-secretion of chemokines alters the immune contexture. Chronic SASP expression leads to the recruitment of immunosuppressive regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) 918. These cells actively suppress cytotoxic T lymphocyte activity, paradoxically protecting senescent cells and nearby pre-malignant cells from immune destruction, thereby fostering an immune-privileged microenvironment 918.
Matrix Remodeling Enzymes and Proteases
Senescent cells actively restructure their physical surroundings by secreting a vast array of extracellular matrix (ECM) modifying enzymes 1417. Matrix metalloproteinases (MMPs), particularly MMP-1 (collagenase-1), MMP-3 (stromelysin-1), and MMP-10 (stromelysin-2), are consistently and heavily upregulated in stress-induced and replicative senescence 151826.
These proteases degrade the structural integrity of the basement membrane and interstitial matrix 1518. While transient MMP expression is critical for wound healing and clearing damaged ECM to allow progenitor cell infiltration, chronic MMP secretion destroys tissue architecture 15. For example, sustained MMP-2 and MMP-9 secretion by senescent fibroblasts directly contributes to pathological lung fibrosis and the loss of dermal elasticity 1527. Furthermore, MMPs act post-translationally to cleave and activate other SASP cytokines and chemokines. They cleave inactive precursors of IL-8 and MCP-1, thereby amplifying the localized inflammatory cascade beyond strict transcriptional control 1824.
In addition to MMPs, senescent cells secrete serine proteases such as urokinase-type plasminogen activator (uPA) and specific regulators such as Plasminogen Activator Inhibitor-1 (PAI-1/SERPINE1) 1826. The dysregulation of these factors contributes heavily to pro-fibrotic conditions and disrupts normal fibrinolysis and vascular health, establishing a physically rigid and dysfunctional tissue bed 1826.
Growth Factors and Soluble Signaling Molecules
Growth factors are central to the regenerative functions of the acute SASP, but they also fuel pathological tissue remodeling when chronically expressed 1417.
- Transforming Growth Factor Beta (TGF-β): A highly pleiotropic cytokine that induces profibrotic responses, stimulates cell migration, and is a potent inducer of secondary, or paracrine, senescence in neighboring epithelial and stromal cells 14.
- Vascular Endothelial Growth Factor (VEGF): Secreted heavily by senescent cells to stimulate angiogenesis. In the tumor microenvironment, this inappropriate angiogenic signaling provides necessary blood supply to expanding neoplastic masses, inadvertently feeding tumor growth 1428.
- Platelet-Derived Growth Factor-AA (PDGF-AA): Highly concentrated in the SASP and identified as an essential molecule for optimal epidermal wound healing and closure, demonstrating the pro-regenerative capacity of transient senescence 214.
- Insulin-like Growth Factor Binding Proteins (IGFBPs): Particularly IGFBP-2, -3, -4, -5, and -7. IGFBP7 is recognized as a dominant factor in transmitting oncogene-induced senescence to neighboring healthy cells, effectively acting as a paracrine emergency brake on tissue-level proliferation 142429.
Non-Protein and Extracellular Vesicle Components
Beyond soluble proteins, the SASP profile includes vital non-proteinaceous components 1017. Damage-Associated Molecular Patterns (DAMPs), particularly High Mobility Group Box 1 (HMGB1), are released from the nucleus of stressed cells, acting as alarmins that attract immune cells and amplify inflammation 18. Senescent cells also release elevated levels of reactive oxygen species (ROS) and nitric oxide, which directly induce oxidative DNA damage in neighboring cells 1018.
They additionally synthesize and release bioactive lipid mediators, such as eicosanoids derived from arachidonic acid. This includes prostaglandin E2 (PGE2) via COX-2 upregulation, and leukotrienes via ALOX5 1824. These lipids act as powerful, highly localized amplifiers of inflammation. Furthermore, senescent cells exhibit altered exosome and ectosome shedding 10. These extracellular vesicles (EVs) contain microRNAs, fragmented DNA, and concentrated SASP proteins, serving as highly protected, long-distance delivery vehicles capable of altering the transcriptomic profiles of distal cells, spreading the senescence phenotype systemically 101217.
Summary of Major SASP Components
| Component Category | Primary Examples | Principal Biological Effects on Microenvironment | Citations |
|---|---|---|---|
| Inflammatory Cytokines | IL-6, IL-1α, IL-1β, TNF-α | Reinforces autocrine senescence; drives systemic inflammaging; impairs adjacent stem cell proliferation and differentiation. | 101417 |
| Chemokines | IL-8 (CXCL8), CCL2 (MCP-1), CXCL1 | Recruits immune cells (macrophages, NK cells) for acute clearance; chronically recruits immunosuppressive Tregs and MDSCs. | 141718 |
| Proteases (MMPs & Serine) | MMP-1, MMP-3, MMP-10, uPA, PAI-1 | Cleaves and activates chemokines; degrades ECM resulting in loss of tissue integrity; promotes tumor invasiveness and metastasis. | 151826 |
| Growth Factors | TGF-β, VEGF, PDGF-AA, IGFBP7 | TGF-β and IGFBP7 induce paracrine senescence; VEGF promotes neoangiogenesis; PDGF-AA drives epidermal wound repair. | 21429 |
| Lipids, DAMPs & Non-Proteins | ROS, HMGB1, PGE2, Leukotrienes, EVs | Induces oxidative stress in adjacent cells; highly amplifies localized inflammation; spreads senescence via circulating extracellular vesicles. | 101824 |
Transcriptional and Epigenetic Regulation of the SASP
The expression of the SASP requires massive transcriptomic and epigenetic reorganization within the senescent cell, controlled by multiple intersecting signaling cascades 2330.
Transcriptional Control and Signaling Pathways
The primary driver of the SASP is the activation of the Nuclear Factor kappa B (NF-κB) transcription factor 219. Upon severe DNA damage or oxidative stress, the DDR activates ATM and CHK2 kinases, which initiate a cascade leading to NF-κB translocation into the nucleus, where it binds to the promoters of core SASP genes, notably IL-6 and IL-8 161819. Inhibition of NF-κB can completely abrogate the inflammatory components of the SASP, bypassing the secretory phenotype while maintaining the cell cycle arrest 1931.
Concurrently, the p38 Mitogen-Activated Protein Kinase (MAPK) and the Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathways act as essential modulators 1232. The JAK/STAT pathway is particularly critical for the downstream signaling of many interleukins; IL-6 signaling through JAK1/2 stabilizes the senescent state and amplifies the inflammatory feedback loop 323334.
Epigenetic and RNA Splicing Mechanisms
Recent advancements highlight the profound role of epigenetics and transcriptomic instability in regulating senescence 3537. The physical clustering of SASP genes (such as the MMPs and CXCLs) on specific chromosomal loci allows for widespread upregulation via broader chromatin remodeling 27. The relocation of histone variants, such as macroH2A1, away from SASP genes during stress responses ensures that the promoters for these massively expressed secreted factors remain physically open and highly transcribed 27.
Furthermore, aging is accompanied by an overall loss of RNA splicing fidelity 36. Research led by Harries and colleagues demonstrates that the dysregulation of RNA splicing factors is a core hallmark of cellular aging 3738. Increased RNA polymerase II elongation rates impair co-transcriptional splicing, and altered expression of upstream transcription factors like FOXO1 and ETV6 directly influences the alternative splicing machinery 3639. This generates mis-spliced mRNA isoforms that reinforce the senescent arrest and the SASP 3639. Notably, interventions utilizing senomorphic compounds, such as the MEK inhibitor trametinib, have been shown to restore splicing homeostasis and partially reverse senescence characteristics in models of progeroid syndromes (like Hutchinson-Gilford Progeria and Cockayne syndrome), offering a novel regulatory node for intervention 3538.
Autocrine and Paracrine Effects on the Microenvironment
The biological consequences of cellular senescence are mediated almost entirely through the complex autocrine and paracrine interactions of the SASP 19. Autocrine signaling operates as a self-sustaining feedback loop. For example, the secretion of IL-6 and its subsequent binding to the cell's own surface receptors actively maintains the senescence-associated cell cycle arrest, ensuring that the cell does not aberrantly re-enter the replication phase despite severe molecular damage 192333.
Induction of Paracrine Senescence
Perhaps the most insidious feature of the SASP is its capacity to induce "secondary" or "paracrine" senescence in adjacent, perfectly healthy cells - a phenomenon often described as a bystander effect 1419. When a primary cell undergoes senescence, its secretion of factors such as TGF-β, IGFBP7, IL-6, and ROS bathes the surrounding microenvironment 14.
Prolonged exposure to these factors triggers DNA damage responses and oxidative stress in neighboring cells, forcing them into a senescent state even if they have not experienced the original intrinsic stressor (e.g., telomere attrition or irradiation) 51019. This paracrine amplification implies that senescence behaves akin to a localized pathogenic infection within the tissue; a small burden of primary senescent cells (sometimes representing merely 2-3% of the total tissue cell population) can precipitate widespread functional collapse through compounding secondary senescence 1040.
Extracellular Matrix Remodeling and Mechanical Softening
While acute ECM degradation via MMPs allows for the clearance of dead tissue, the chronic SASP fundamentally alters tissue biomechanics 1526. Senescent fibroblasts upregulate fibrillar collagens and fibronectin, attempting to rebuild the matrix 1418. However, combined with simultaneous, dysregulated MMP secretion, the result is severe architectural disorganization 1841.
Research assessing the biomechanical effects of senescent fibroblasts reveals that their presence correlates with structural reorganization of the collagen network, characterized by increased branching and mechanical softening 41. This altered matrix impairs cellular adhesion and migration, contributing directly to tissue fragility. In aging skin and cardiovascular tissue, this SASP-mediated fibrosis drastically reduces elasticity and compliance, leading directly to macroscopic functional impairment 1541.
Influence of Senescence Triggers on Secretory Profiles
The specific composition and magnitude of the SASP are highly dependent upon the molecular trigger that initiated the senescence program 81014. Not all senescence is equal, and the variations have profound implications for clinical interventions 5.
Replicative Senescence
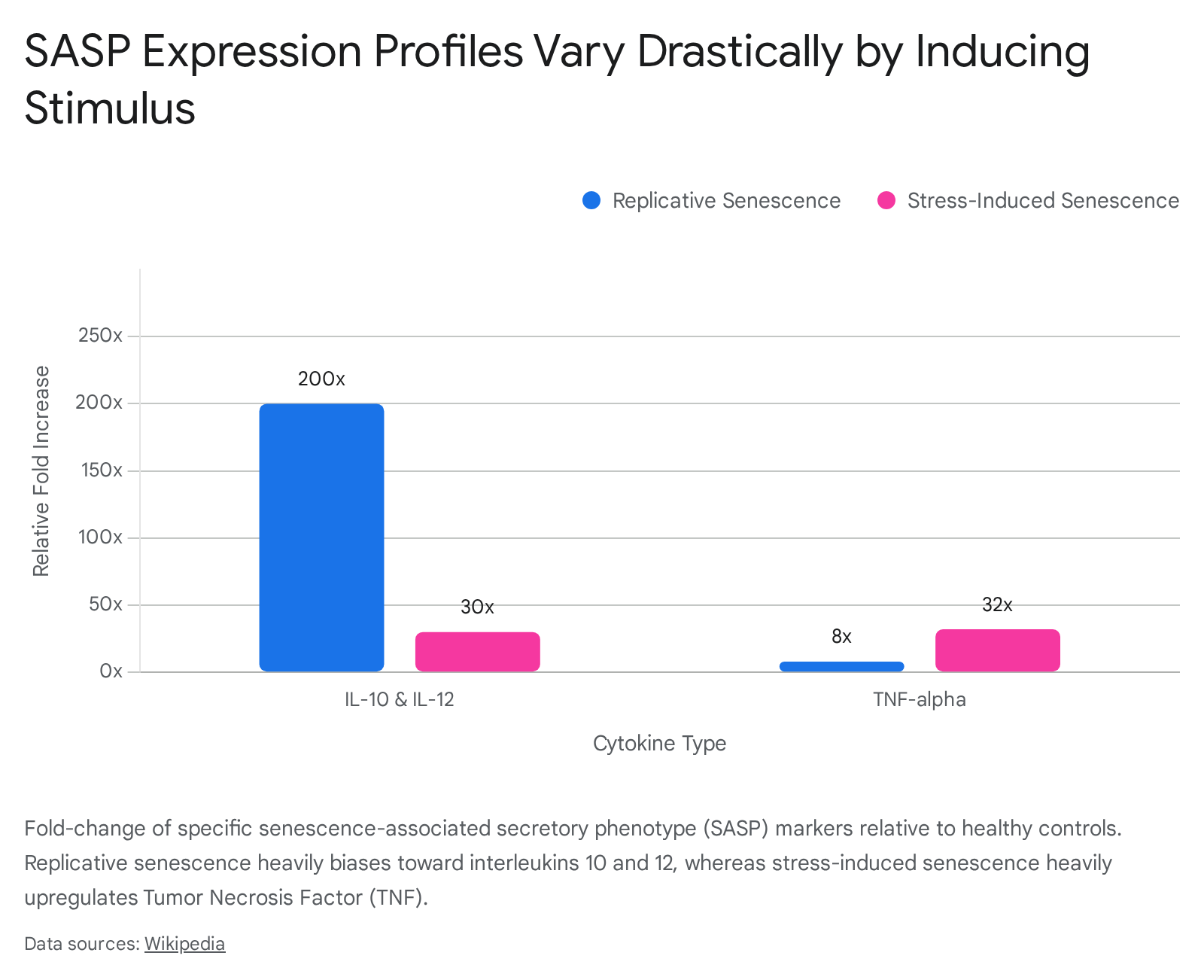
Replicative senescence occurs when somatic cells reach their Hayflick limit due to progressive telomere shortening over successive divisions 48. The eroded telomeres are recognized by the cellular machinery as unrepairable double-strand DNA breaks, triggering a persistent DDR and subsequent p53-mediated arrest 89. The SASP resulting from replicative senescence tends to feature a specific subset of inflammatory markers. For instance, transcriptomic analyses indicate that anti-inflammatory and immunoregulatory cytokines such as IL-12 and IL-10 can be upregulated more than 200-fold in replicatively senescent cells, a stark contrast to stress-induced senescence where these interleukins rise minimally (usually under 30-fold) 10. Conversely, Tumor Necrosis Factor (TNF) increases drastically in stress-induced senescence but only marginally in replicative scenarios 10.
Oncogene-Induced Senescence
Oncogene-induced senescence (OIS) is an acute, hyper-proliferative stress response 816. When a powerful oncogene, such as mutant RAS or BRAF, is activated in a normal cell, it causes massive replication stress, depletion of nucleotide pools (via downregulation of RRM2), and widespread genomic instability 9. The cell senses this catastrophic oncogenic drive and forcibly halts proliferation 89.
Because OIS is fundamentally a tumor-suppressive fail-safe, its associated SASP is typically much more intense and highly enriched in pro-inflammatory, immune-recruiting chemokines (like IL-6 and IL-8) compared to replicative senescence 4910. The immediate evolutionary goal of the OIS SASP is to flag the pre-malignant cell for rapid destruction by the immune system before it can bypass the senescence barrier and become a full-blown malignancy 4818.
Therapy-Induced Senescence
Therapy-induced senescence (TIS) arises when cancer cells or surrounding healthy stroma are exposed to massive genotoxic insults from clinical interventions, such as ionizing radiation or systemic chemotherapy (e.g., doxorubicin, cisplatin) 8916. While these therapies successfully halt primary tumor growth by inducing widespread DNA damage and forcing cells into senescence, they inadvertently generate a highly pathological SASP within the tumor microenvironment 816.
The TIS SASP is particularly problematic in oncology. The prolonged secretion of survival factors, VEGF, and MMPs by therapy-induced senescent cells remodels the microenvironment to be more favorable for the survival and eventual metastasis of any remaining, non-senescent cancer cells 8916. Furthermore, TIS directly upregulates immune checkpoint molecules like PD-L1 on senescent cells, dampening anti-tumor immunity, promoting immune evasion, and driving severe therapeutic resistance 9.
Summary of SASP Variations by Inducer
| Senescence Trigger | Primary Molecular Driver | Distinguishing SASP Characteristics | Clinical & Physiological Impact |
|---|---|---|---|
| Replicative Senescence (RS) | Progressive telomere attrition; standard DNA damage response. | Moderate overall SASP intensity; highly specific elevation of IL-10 and IL-12 (up to 200-fold) compared to stress-induced variants. | Gradual onset associated with chronological aging and widespread slow tissue degradation. 810 |
| Oncogene-Induced Senescence (OIS) | Hyper-activation of oncogenes (RAS, BRAF); intense replication stress and nucleotide depletion. | Highly robust, intense pro-inflammatory secretome; massive upregulation of IL-6, IL-8, and IGFBP7. | Acts as a potent fail-safe against cancer; intense immune signaling designed for rapid clearance of pre-malignant cells. 8916 |
| Therapy-Induced Senescence (TIS) | Iatrogenic genotoxic stress (chemotherapy, radiation) causing sudden, massive DNA breaks. | Paracrine signaling heavily skewed toward pro-survival factors, VEGF, and matrix remodeling MMPs; upregulation of PD-L1. | Drives chemo-resistance, tumor recurrence, and late-stage metastasis by providing a supportive niche for surviving cancer cells. 8916 |
The Dual Role of Senescence in Physiology and Pathology
The evolutionary preservation of cellular senescence across species implies that the SASP must offer critical survival advantages, at least during early development and peak reproductive years 710.
Acute Secretion in Embryogenesis and Tissue Repair
In transient, controlled environments, senescent cells and their SASP are physiologically essential 715. During embryogenesis, specific populations of cells undergo programmed senescence to guide tissue patterning, structural morphogenic signaling, and organogenesis. The tightly orchestrated SASP allows for spatial patterning and is rapidly cleared by macrophages via programmed cell clearance once structural development is complete, ensuring proper organ formation 215.
Similarly, in adult organisms, the SASP is vital for optimal wound healing and tissue regeneration 1420. Upon severe injury, fibroblasts differentiate into myofibroblasts, which eventually senesce to prevent excessive scarring 2. These acute senescent cells secrete high levels of PDGF-AA, which stimulates the migration and proliferation of surrounding epithelial cells to rapidly close the wound 214. Furthermore, the transient burst of SASP MMPs breaks down fibrotic scar tissue, and the subsequent rapid immune-mediated clearance of these senescent cells resolves the injury site, restoring full tissue homeostasis 215.
Chronic Secretion in Aging and Degenerative Disease
The pathology of the SASP arises when the immune system fails to clear senescent cells, allowing their acute repair signals to become chronic and deleterious 715. As organisms age, senescent cells accumulate exponentially in various tissues 2231. The continuous secretion of IL-6, IL-8, and matrix-degrading proteases shifts the local environment into a state of chronic, sterile inflammation 1522.
This localized inflammation exhausts tissue-resident stem cells, preventing normal renewal. In the skin, this manifests as thinning and loss of elasticity; in the lungs, it drives fibrotic pathologies; and systemically, the leakage of SASP factors into the bloodstream exacerbates metabolic syndromes, cardiovascular deterioration, and generalized physical frailty 152842.
Paradoxical Effects in the Tumor Microenvironment
The relationship between the SASP and cancer is perhaps the most heavily studied paradox in geroscience 1618. Early in life, senescence operates as a potent tumor suppressor 810. When a cell suffers an oncogenic mutation, senescence permanently arrests its growth, and the resulting acute SASP recruits NK cells and macrophages to eradicate the premalignant threat before it can establish a tumor 818.
However, in older individuals harboring established tumors, or in tissue environments subjected to a chronic SASP, senescent cells actively fuel cancer progression 1016. Prolonged exposure to SASP factors like IL-6, IL-8, and TGF-β induces Epithelial-Mesenchymal Transition (EMT) in neighboring non-senescent premalignant cells 81618. EMT is a critical biological shift wherein epithelial cells lose their cell-to-cell adhesion and gain migratory, invasive properties, facilitating metastasis 816. Additionally, the secretion of VEGF promotes tumor neoangiogenesis, while the degradation of the ECM by MMPs clears a physical path for invading cancer cells 918. Consequently, while senescence suppresses cancer strictly cell-autonomously, its SASP heavily promotes cancer cell-non-autonomously 81016.
Cellular Senescence in Organ-Specific Pathologies
The accumulation of SASP-expressing cells drives highly specific pathologies depending on the tissue niche and specific cell subset in which they reside 3141.
Cardiovascular Remodeling and Disease
In the cardiovascular system, cellular senescence plays an unrecognized but pivotal role in cardiac remodeling, fibrosis, and eventual heart failure 1932. Senescent endothelial cells secrete SASP factors such as Angiotensin II (ANG II) and Endothelin-1 (ET-1), which directly induce senescence in neighboring cardiomyocytes 19. Senescent cardiomyocytes exhibit reduced contractile ability, elevated pacing frequency, and impaired shortening 32.
In conditions such as myocardial infarction or septic cardiomyopathy, the localized secretion of CXCL10 and CCL5 exacerbates immune infiltration and maladaptive fibrotic remodeling 1932. Notably, researchers utilizing advanced single-cell RNA sequencing in transgenic mouse models (such as the p16-CreERT2-tdTomato reporter mice) have demonstrated that specific subsets of p16-positive fibroblasts heavily upregulate TGF-β and BMP4 4143. This activation promotes the expression of collagen genes (Col4a1 and Col5a3), driving age-associated cardiac fibrosis. Selective elimination of these p16-positive fibroblasts has been shown to ameliorate cardiac fibrosis, restoring tissue properties to levels comparable to young models 41.
Neurological and Cognitive Decline
The central nervous system is highly vulnerable to the inflammatory elements of the SASP, lacking robust regenerative capacity to counteract structural damage 4445. Brain aging and neurodegenerative disorders, including Alzheimer's disease, are tightly linked to the accumulation of senescent glial cells and microglia 194446. Senescent astrocytes downregulate critical glutamate transporters, leading to excitotoxicity and neuronal death, while simultaneously releasing massive quantities of IL-6 and TGF-β 8. The resulting severe neuro-inflammation disrupts synaptic plasticity, exacerbates amyloid-beta pathology, and induces tau hyperphosphorylation, driving severe cognitive decline 194446.
Metabolic Dysfunction and Adipose Tissue
Adipose tissue is highly susceptible to cellular senescence, particularly in the context of obesity, which effectively accelerates biological aging within the fat deposits 4246. Senescent preadipocytes and fat cell progenitors accumulate in adipose deposits, secreting abnormally high levels of TNF-α, IL-6, and MCP-1 3342. This localized SASP impedes normal adipocyte differentiation, leading to lipid storage dysfunction, hypertrophic adipocyte formation, and severe insulin resistance 102546. Furthermore, systemic leakage of these adipocyte-derived inflammatory factors promotes the premature aging of circulating T cells, expanding senescent T cell subsets and linking metabolic dysfunction directly to systemic immunosenescence 2542.
Therapeutic Interventions Targeting Senescence
Recognizing that the SASP is a "root cause" of multiple age-related morbidities, the field of geroscience has heavily focused on developing pharmacological agents to mitigate its effects 1347. These agents, broadly termed senotherapeutics, are primarily divided into two functional classes: senolytics, which eliminate the cells, and senomorphics, which silence their secretions 121622.
Senolytic Approaches to Cell Clearance
Senolytic drugs selectively induce apoptosis in senescent cells by targeting and inhibiting their uniquely upregulated Senescent Cell Anti-Apoptotic Pathways (SCAPs) 121348. By physically eliminating the senescent cell, senolytics permanently eradicate the source of the pathological SASP, allowing tissue progenitor cells to repopulate the niche 102249.
The most prominent first-generation senolytic regimen is the combination of Dasatinib (a tyrosine kinase inhibitor approved for leukemia) and Quercetin (a naturally occurring flavonoid) (D+Q) 424650. Dasatinib targets the ephrin dependence of senescent cells, while quercetin broadly inhibits BCL-xL and other specific survival factors 44. In preclinical models, D+Q dramatically reduces senescent cell burden, improves physical function, and alleviates cardiovascular and pulmonary dysfunction 1351. Other notable senolytics include Navitoclax (a potent BCL-2/BCL-xL inhibitor) and Fisetin, which modulates PI3K/AKT/mTOR apoptosis pathways 324252.
Recent developments also highlight targeted metabolic interventions. Research indicates that inhibiting glutaminase 1 (GLS1) effectively targets senescent cell metabolism, resulting in specific senolysis and the alleviation of age-related physiological decline in varied tissue models 4353.
Senomorphic Suppression of Secretory Pathways
Unlike senolytics, senomorphics (also referred to as senostatics) do not kill the senescent cell; instead, they intervene in the intracellular signaling pathways (such as mTOR, NF-κB, or JAK/STAT) to suppress the transcription and secretion of the SASP 121622. This approach mitigates the inflammatory toxicity of the cell without running the risk of massive cellular die-off or immediate structural tissue collapse in highly senescent organs 214454.
- mTOR Inhibitors (Rapamycin): Rapamycin is one of the most robustly validated geroprotectors in mammalian models 2251. By inhibiting the mechanistic target of rapamycin (mTOR), rapamycin dramatically suppresses the translation of key SASP factors, reduces oxidative stress, and extends both healthspan and maximum lifespan in mice 224650.
- NF-κB Modulators (Metformin): Metformin, a highly prevalent first-line therapeutic for Type 2 Diabetes, exerts potent senomorphic effects primarily by inhibiting NF-κB and dampening metabolic stress, thereby reducing the systemic release of SASP cytokines 223151.
- JAK/STAT Inhibitors (Ruxolitinib): Ruxolitinib, an FDA-approved inhibitor of JAK1 and JAK2 kinases, used primarily for myelofibrosis and polycythemia vera, has emerged as a powerful senomorphic 314055. By blocking the JAK/STAT signaling pathway, ruxolitinib severely curtails the production and localized reinforcement of interleukins within the SASP 3233. In aged mouse models, ruxolitinib treatment dramatically reduces systemic inflammation, reverses adipose tissue dysfunction, and rescues bone regenerative capacity to youthful levels 223233. It has also demonstrated efficacy in rescuing progerin-induced cell cycle arrest and bone fractures in models of Hutchinson-Gilford Progeria Syndrome 56.
Clinical Trials of Senotherapeutics
The translation of senotherapeutics from animal models to human clinical trials is progressing rapidly, aiming to evaluate safety, target engagement, and clinical efficacy across numerous specific age-related indications 455152.
- Senolytics in Humans: Early Phase 1/Phase 2 pilot studies of D+Q have shown promising results. A landmark first-in-human trial involving patients with Idiopathic Pulmonary Fibrosis (IPF) - a lethal, senescence-driven fibrotic lung disease - demonstrated significant improvements in objective mobility measures, such as walking distance and speed 135257. Subsequent trials have shown D+Q reduces senescent cell burden in the adipose tissue of diabetic kidney disease patients and holds promise for slowing cognitive decline 315258. Notably, the SToMP-AD trial is evaluating the safety and feasibility of D+Q in older adults with amnestic mild cognitive impairment or early-stage Alzheimer's disease who are tau PET positive 5259.
- Senomorphics in Humans: The Targeting Aging with Metformin (TAME) trial represents a paradigm shift, actively studying thousands of non-diabetic older adults to determine if continuous metformin administration can delay the composite onset of age-related diseases (cardiovascular disease, cancer, and cognitive decline) 315051. Similarly, rapamycin is undergoing evaluation in the PEARL trial to assess improvements in lean muscle mass and immune resilience 5860.
- Advanced Trials of Ruxolitinib: Ruxolitinib is undergoing extensive evaluation beyond its traditional oncology applications. New formulations, such as a once-daily extended-release (XR) tablet, have demonstrated bioequivalence to immediate-release variants in Phase 1 trials (NCT06555081), improving patient compliance 406162. Furthermore, combinations of ruxolitinib with novel agents - such as bomedemstat (an LSD1 inhibitor) and elritercept (a TGF-β receptor ligand trap) - are showing robust efficacy in advanced myelofibrosis, specifically addressing treatment-induced anemia 55. Phase II trials have also demonstrated its strong efficacy as salvage therapy for corticosteroid-refractory sclerotic chronic graft-versus-host disease (cGVHD), directly leveraging its potent anti-fibrotic and SASP-suppressive capabilities to reduce daily corticosteroid dependence 6364. However, real-world data indicates that proactive monitoring is essential, as patients aged 65 and older exhibit higher rates of ruxolitinib discontinuation due to drug-related cytopenias 6566.
| Drug / Intervention | Classification | Primary Molecular Target | Highlighted Clinical Trial Indications / Applications | Citations |
|---|---|---|---|---|
| Dasatinib + Quercetin | Senolytic | Ephrin dependence & BCL-xL anti-apoptotic pathways | Idiopathic Pulmonary Fibrosis (IPF); Diabetic Kidney Disease; early-stage Alzheimer's Disease (SToMP-AD). | 3151525759 |
| Fisetin | Senolytic | PI3K/AKT/mTOR and apoptosis modulation | Osteoarthritis; Frailty; systemic senescent cell clearance in the elderly. | 13424852 |
| Metformin | Senomorphic | AMPK activation; NF-κB pathway inhibition | TAME Trial (delaying multimorbidity); systemic metabolic dysregulation. | 2231505167 |
| Rapamycin | Senomorphic | mTOR kinase inhibition | PEARL Trial (lean muscle mass/longevity); localized reduction of SASP in immune cells. | 224650515860 |
| Ruxolitinib | Senomorphic | JAK1 and JAK2 kinase inhibition | Myelofibrosis (MF); Polycythemia Vera (PV); Sclerotic cGVHD; Cachexia (RUXexia Trial). | 223132335561636468 |
Translational Bottlenecks and Advanced Targeting
Despite immense preclinical promise, transitioning senotherapeutics into standard clinical practice requires navigating substantial biological, pharmacological, and diagnostic hurdles 44454654.
Systemic Immunosuppression and Off-Target Toxicity
The primary limitation of systemic senomorphic administration is the potential for profound off-target immunosuppression 374546. The signaling pathways that regulate the SASP - notably mTOR, NF-κB, and JAK/STAT - are not exclusive to senescent cells; they are absolutely critical for normal immune function, wound healing, and pathogen defense in healthy tissues 153746.
Continuous administration of rapamycin or broad-spectrum JAK inhibitors like ruxolitinib can induce systemic immunodeficiency, glucose intolerance, and delayed tissue repair, side-effects that actively contradict their pro-longevity goals 153746. Similarly, the unselective clearance of cells by senolytics can disrupt beneficial homeostatic functions, particularly since transient SASP expression is required for acute vascular repair, liver regeneration, and maintaining endothelial integrity 21545.
To bypass these bottlenecks, current research is heavily focused on developing precision delivery mechanisms 123749. Novel alternatives include engineering Chimeric Antigen Receptor (CAR) T-cells specifically targeted to unique senescent surface markers. For example, CAR-T cells targeting the urokinase-type plasminogen activator receptor (uPAR) have shown remarkable efficacy in reducing senescent burden in pulmonary and hepatic fibrosis models without the broad toxicity of systemic drugs 102246. Additionally, nanoparticle-based delivery systems and specific antibody-drug conjugates are being developed to restrict senolytic toxicity exclusively to the senescent niche, preventing healthy-cell collateral damage 123749. Finally, "hit-and-run" dosing strategies - where senolytics are administered intermittently (e.g., once weekly or monthly) rather than continuously - are proving highly effective at clearing senescent populations while allowing normal tissue intervals to recover, drastically reducing off-target toxicity profiles 1358.
Biomarker Validation and Clinical Endpoints
A major barrier to regulatory approval of anti-aging therapies is the absence of universally validated, high-throughput biomarkers for senescence in living patients 12214445. While p16INK4a expression and senescence-associated beta-galactosidase (SA-β-gal) are reliable in in vitro assays and biopsy samples, assessing total systemic senescent burden safely and repeatedly is exceedingly difficult 22122.
Current efforts in geroscience emphasize the use of composite circulating SASP scores 112831. By analyzing specific panels of chemokines, growth factors, and proteases in patient blood, researchers can infer the underlying senescent cell load 28. These circulating markers strongly correlate with objective clinical outcomes such as grip strength, gait speed, and overall intrinsic capacity 284769. Integrating these biomarker panels into clinical trial designs - as heavily recommended by the Intrinsic Capacity, Frailty and Sarcopenia Research (ICFSR) Task Force - is essential for proving target engagement and facilitating the eventual regulatory recognition of "aging" and "frailty" as directly treatable medical indications 4769.
By successfully modulating the Senescence-Associated Secretory Phenotype through highly targeted, biomarker-driven interventions, modern medicine stands on the precipice of shifting from reactive single-disease management to proactive healthspan extension, fundamentally treating the biological root of chronic disease.