The role of the mTOR pathway in aging
Introduction
Biological aging is fundamentally characterized by the gradual, progressive decline in physiological integrity, leading to impaired tissue function, loss of homeostatic capacity, and exponentially increased vulnerability to age-related pathologies such as neurodegeneration, cardiovascular disease, metabolic syndrome, and neoplastic diseases 123. Central to the molecular understanding of aging is the paradigm that the deterioration of cellular homeostasis is not merely a passive accumulation of entropy, but rather a process actively regulated by highly conserved metabolic signaling networks. Among these regulatory systems, the mechanistic target of rapamycin (mTOR) pathway functions as a primary integrating node, coordinating cellular metabolism, growth, and survival in response to diverse environmental inputs, including nutrient availability, energy status, and growth factors 245.
The mTOR protein is a 289-kDa atypical cytoplasmic serine/threonine kinase that belongs to the phosphoinositide 3-kinase (PI3K)-related kinase (PIKK) family 56. While the baseline activity of the mTOR pathway is essential for development and physiological maintenance, the chronic hyperactivation of mTOR signaling in post-mitotic tissues is intrinsically linked to the acceleration of aging mechanisms. Persistent anabolic signaling drives cellular hypertrophy, replicative senescence, and chronic sterile inflammation - collectively recognized as primary hallmarks of biological aging 78.
Consequently, the pharmacological modulation of the mTOR pathway has become a central focus of experimental gerontology. The inhibition of mTOR by rapamycin (sirolimus) and its synthetic analogs (rapalogs) is currently recognized as the most robust and reproducible pharmacological intervention capable of extending both median and maximal lifespan across diverse eukaryotic model organisms, ranging from Saccharomyces cerevisiae and Caenorhabditis elegans to Drosophila melanogaster and Mus musculus 12910. However, translating the geroprotective efficacy of rapamycin from laboratory models to broad clinical application in healthy human adults is complicated by the pleiotropic nature of the mTOR pathway. Developing optimal therapeutic protocols requires a rigorous understanding of the distinct multiprotein complexes formed by mTOR, their specific downstream biochemical cascades, and the profound differences between continuous and intermittent pharmacological inhibition.
Historical Discovery of Rapamycin
The discovery of rapamycin represents a remarkable trajectory in natural product pharmacology, originating from efforts to investigate localized immunological phenomena in an isolated ecosystem. In November 1964, a Canadian-led scientific initiative known as the Medical Expedition to Easter Island (METEI) embarked on the Royal Canadian Navy's H.M.C.S. Chief Scott, traveling to Rapa Nui (Easter Island), a highly remote volcanic island situated 2,200 kilometers from the nearest inhabited landmass in the South Pacific 111213.
Among the international team of physicians and researchers was microbiologist Georges Nógrády, whose primary research objective was to determine why the local indigenous population exhibited an unexpected resistance to tetanus 1314. Despite a high prevalence of horses on the island and the local custom of walking barefoot, tetanus infections were exceptionally rare 13. To investigate this anomaly, Nógrády collected systematic soil samples from various microenvironments across the island, including unique samples from the crater of Rano Kau, an extinct volcano 1314.
Although the specific investigation into tetanus spores yielded inconclusive results regarding the population's immunity, Nógrády preserved the soil samples and subsequently transferred them to researchers at Ayerst Pharmaceuticals (which later transitioned to Wyeth-Ayerst and eventually Pfizer) in Montreal for systematic antimicrobial screening 1213. Within these samples, the Ayerst research team, spearheaded by Dr. Surendra Sehgal, isolated a novel aerobic Gram-positive soil bacterium, designated Streptomyces hygroscopicus AY B-994 [NRRL 5491] 1112. From the mycelium of this bacterium, the team extracted a secondary metabolite using organic solvents. In direct homage to the island's indigenous Polynesian name, Rapa Nui, the isolated macrolide compound was officially named rapamycin 121314.
Initial pharmacological characterization in the early 1970s revealed that rapamycin possessed potent broad-spectrum anti-fungal properties, successfully inhibiting the proliferation of pathogenic fungi such as Candida albicans, Microsporum gypseum, and Trichophyton granulosum 1113. However, its clinical development as a standard antifungal agent was abruptly halted when subsequent in vivo testing revealed that the compound induced profound immunosuppressive activity, rendering it unsuitable for treating routine fungal infections 1314.
Recognizing that the compound possessed unique biological properties beyond simple antifungal activity, Sehgal distributed samples of Streptomyces hygroscopicus to the U.S. National Cancer Institute for further screening 13. Subsequent assays in the 1980s demonstrated that rapamycin effectively inhibited the in vitro cell growth of various mammalian tumor cell lines 111215. The critical observation that a single macrolide compound could arrest the proliferation of both single-celled fungi and complex human immune and neoplastic cells suggested that rapamycin targeted an evolutionarily conserved, fundamental regulatory mechanism governing cellular growth 12.
This realization precipitated a surge of molecular studies in the early 1990s, leading to the successful identification of the target of rapamycin (TOR) kinase in yeast, and shortly thereafter, the discovery of the highly conserved mammalian ortholog (mTOR) 512. By 1999, following extensive clinical trials, the United States Food and Drug Administration (FDA) approved the use of rapamycin (marketed under the generic name sirolimus) as a primary immunosuppressant designed to prevent allograft rejection in renal transplant patients 1416. It was not until a decade later, in 2009, that a seminal investigation published in Nature formally demonstrated that rapamycin administration could robustly extend the lifespan of genetically heterogeneous mice, officially anchoring the molecule as a cornerstone of modern geroscience 141616.
Structural Biology of the mTOR Complexes
The mTOR kinase is not a solitary effector; rather, it functions exclusively as the catalytic core of two structurally distinct and functionally divergent multiprotein signaling complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) 181718. The distinct molecular architecture of these complexes - specifically their unique scaffold and regulatory subunits - dictates their specific upstream inputs, substrate affinities, subcellular localizations, and, crucially, their differential sensitivities to pharmacological inhibition by rapamycin 619.
Architecture and Function of mTOR Complex 1 (mTORC1)
mTORC1 operates as the primary cellular hub for nutrient and energy sensing. High-resolution cryo-electron microscopy structural studies have determined that mTORC1 forms an obligate dimer 18. The complex is defined by the interaction of the mTOR protein kinase with the essential scaffold protein Raptor (regulatory-associated protein of mTOR) 1819. Raptor functions as the critical identifying subunit that recruits specific downstream substrates to the mTOR kinase domain for phosphorylation 20. mTORC1 also requires the incorporation of the 40 kDa proline-rich Akt substrate (PRAS40), which acts as a regulatory inhibitor 518.
Both mTOR complexes share certain foundational components. mTORC1 includes mLST8 (mammalian lethal with Sec13 protein 8, also known as GβL), which is absolutely required for complex assembly and the stabilization of the kinase loop, as well as the inhibitory regulatory protein DEPTOR (DEP domain-containing mTOR-interacting protein) 518. Additionally, the proteins Tel2 and Tti1 transiently associate with the complex to facilitate initial assembly 18.
Functionally, mTORC1 coordinates anabolic cellular processes. It is activated by a convergence of favorable environmental inputs, including high intracellular amino acid concentrations, robust energy status (high ATP/AMP ratio), oxygen availability, and growth factor stimulation 521. Upon activation, mTORC1 promotes protein synthesis, lipid biogenesis, and nucleotide synthesis while concurrently suppressing catabolic processes, most notably autophagy and lysosomal biogenesis 517.
A defining pharmacological characteristic of mTORC1 is its acute vulnerability to rapamycin. Rapamycin does not act as a traditional competitive kinase inhibitor that directly binds to the ATP-binding pocket of the active site. Instead, upon entering the cytoplasm, rapamycin binds to the intracellular immunophilin FKBP12 (FK506-binding protein 12) 519. This newly formed gain-of-function complex, rapamycin-FKBP12, subsequently docks onto a highly specific domain on the mTOR protein known as the FRB (FKBP12-rapamycin binding) domain 2122. Structural analysis indicates that the binding of rapamycin-FKBP12 adjacent to the active site induces steric hindrance, physically occluding Raptor from recruiting its specific substrates, thereby arresting mTORC1-mediated downstream signaling 1822. Furthermore, research has indicated that phosphatidic acid (PA), a metabolic product of phospholipase D (PLD), is required for the stabilization of both mTOR complexes and competes directly with rapamycin for interaction with the FRB domain 1819.
Architecture and Function of mTOR Complex 2 (mTORC2)
In contrast to the nutrient-sensing role of mTORC1, mTORC2 functions primarily as a major effector node downstream of insulin, insulin-like growth factor 1 (IGF-1), and other growth factor receptor signaling pathways 1821. Its primary physiological roles include the regulation of cell survival, global cellular metabolism, cellular proliferation, and the spatial reorganization of the actin cytoskeleton 521.
mTORC2 is defined by the interaction of the mTOR kinase with its unique scaffold protein, Rictor (rapamycin-insensitive companion of mTOR) 1819. The complex utilizes several specialized subunits not found in mTORC1, including mSin1 (mammalian stress-activated protein kinase-interacting protein 1) and Protor-1/2 518. Like mTORC1, mTORC2 incorporates the shared subunits mLST8 and DEPTOR 518.
The defining pharmacological feature of a fully assembled mTORC2 complex is its acute insensitivity to rapamycin 1921. Cryo-electron microscopy of mTORC2 has revealed the structural basis for this resistance. The specific subunit mSin1 binds to the mTOR/Rictor complex in a manner that spatially barricades the FRB domain and the active site 22. Consequently, the rapamycin-FKBP12 complex is physically prevented from docking onto pre-existing, intact mTORC2 structures 2122.
However, mTORC2 is not entirely immune to the drug. While acute exposure leaves the complex unaffected, chronic or prolonged administration of rapamycin eventually depletes mTORC2 functionality 1819. Because rapamycin-FKBP12 readily binds to the pool of free, unassembled mTOR monomers in the cytoplasm, it effectively halts the de novo assembly of new mTORC2 complexes. Over the course of 24 to 48 hours, as existing mTORC2 complexes naturally degrade and are not replaced, total mTORC2 signaling capacity is severely attenuated 1821.
The fundamental differences between these two regulatory nodes are summarized below.
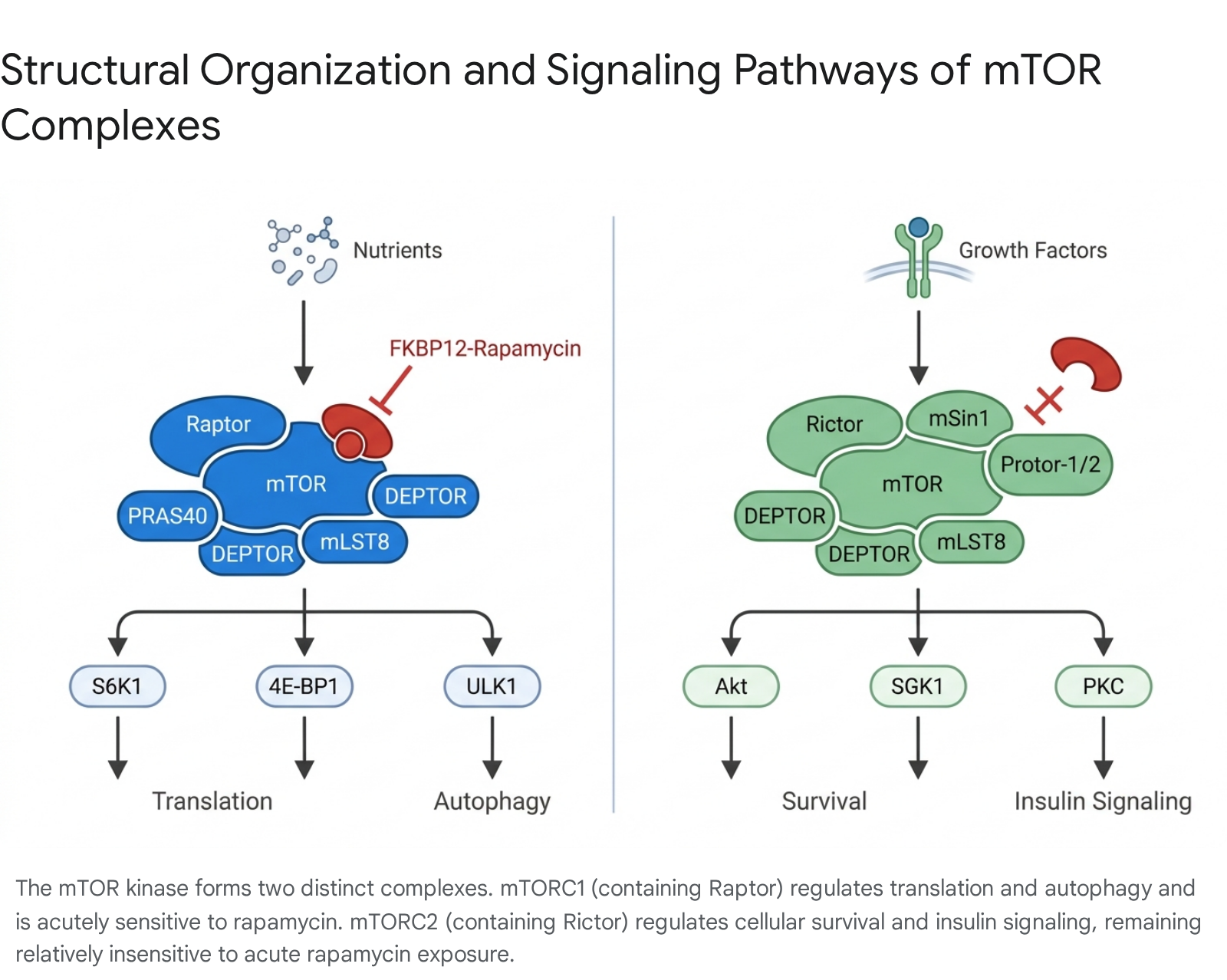
| Feature | mTOR Complex 1 (mTORC1) | mTOR Complex 2 (mTORC2) |
|---|---|---|
| Defining Scaffold Protein | Raptor | Rictor |
| Unique Complex Subunits | PRAS40 | mSin1, Protor-1/2 |
| Shared Complex Subunits | mLST8, DEPTOR | mLST8, DEPTOR |
| Primary Activating Stimuli | Amino acids (leucine, arginine), ATP/AMP ratio, Growth factors, Oxygen | Insulin, IGF-1, Growth factor receptors, G-protein coupled receptors |
| Primary Physiological Roles | mRNA translation, lipid synthesis, ribosomal biogenesis, autophagy inhibition | Cellular survival, Akt activation, actin cytoskeleton organization |
| Key Downstream Targets | S6K1, 4E-BP1, ULK1, LARP1 | Akt (S473, T450, S477), SGK1, PKC family (alpha, delta, epsilon), IRS1 |
| Sensitivity to Rapamycin | Acutely sensitive (immediate inhibition via FKBP12-rapamycin docking) | Acutely insensitive; inhibited only by chronic exposure preventing de novo assembly |
Downstream Biochemical Cascades
The profound influence of the mTOR pathway on the aging process is not an abstract phenomenon; it is executed through precise biochemical cascades that dictate the balance between cellular construction and cellular demolition. The chronic hyperfunction of these downstream pathways in late life contributes to macromolecular damage, senescence, and the exhaustion of stem cell reserves 17.
Regulation of mRNA Translation and Cell Growth
When favorable environmental conditions trigger the activation of mTORC1, the complex exerts dominant control over protein synthesis primarily by phosphorylating two critical target families: ribosomal protein S6 kinase (S6K) and the eukaryotic translation initiation factor 4E-binding proteins (4E-BPs) 2023.
Upon recruitment by Raptor, mTORC1 phosphorylates S6K1 at a specific threonine residue (T389) 2324. This phosphorylation event is a prerequisite for the full activation of S6K1, which also requires subsequent phosphorylation at T229 by phosphoinositide-dependent kinase-1 (PDK1) 23. Once fully active, S6K1 phosphorylates numerous targets involved in the initiation and elongation phases of mRNA translation, driving ribosomal biogenesis and promoting an increase in overall cell size (hypertrophy) 23.
Concurrently, mTORC1 phosphorylates 4E-BP1 at multiple functional sites 2027. In its unphosphorylated, baseline state, 4E-BP1 acts as a translational repressor by tightly binding to the initiation factor eIF4E, preventing it from interacting with the translational machinery. Phosphorylation by mTORC1 induces a structural conformational change that forces 4E-BP1 to release eIF4E. The liberated eIF4E is then free to incorporate into the multi-subunit eIF4F complex at the 5' cap of messenger RNAs, effectively initiating cap-dependent translation 23. While both S6K and 4E-BP pathways promote protein synthesis, experimental evidence indicates they regulate distinct cellular parameters: S6K primarily controls cell size without affecting cell cycle progression, whereas 4E-BP actively controls cell proliferation and cycle progression without independently altering cell size 23.
The intrinsic kinase activity of mTOR itself is also highly dynamic and regulated by autophosphorylation. Advanced mass spectrometry analyses have identified novel phosphorylation events on S2159 and T2164 within the mTOR kinase domain 25. This dual phosphorylation cooperatively modulates the interaction between mTOR, Raptor, and PRAS40, augmenting the intrinsic kinase activity of mTORC1 and hyper-driving its signaling to S6K1 and 4E-BP1, thereby accelerating cellular proliferation 25.
Regulation of Autophagy and the ULK1 Feedback Loop
Autophagy is an essential, highly conserved cellular quality-control mechanism involving the lysosomal degradation and recycling of damaged organelles, misfolded proteins, and lipid droplets. The efficiency of autophagic clearance naturally declines with advancing age, contributing to the accumulation of toxic cellular debris associated with neurodegeneration and cardiac dysfunction 1729.
mTORC1 acts as the master negative regulator of this process. Under nutrient-replete conditions, active mTORC1 suppresses catabolism by directly phosphorylating and inactivating ULK1 (Unc-51-like kinase 1), the primary initiator kinase of the autophagic cascade 2026. Concurrently, mTORC1 phosphorylates the transcription factor EB (TFEB), restricting it to the cytoplasm and preventing the transcription of genes required for lysosome biogenesis 15.
Conversely, when mTORC1 is inhibited - either through nutrient deprivation or the administration of rapamycin - the repressive phosphorylation on ULK1 is removed. The activated ULK1 complex (which includes ATG13 and FIP200) initiates the formation of autophagosomes to begin cellular recycling 2029.
Crucially, emerging biological research has revealed that the relationship between mTORC1 and ULK1 is not strictly unidirectional; it operates as a sophisticated, mutually antagonistic feedback loop 2026. Active ULK1 does not merely initiate autophagy; it actively fortifies its own activation by directly attacking mTORC1. Upon activation, ULK1 induces multi-site phosphorylation on the mTORC1 scaffold protein, Raptor, heavily phosphorylating specific serine residues (Ser855, Ser859, and moderately Ser792) 2026. This targeted phosphorylation physically alters Raptor, creating a state of steric hindrance that prevents substrate docking. As a result, even if upstream growth signals are present, the phosphorylated Raptor cannot bind to S6K1 or 4E-BP1 2026. This novel negative feedback loop ensures a decisive, binary cellular switch, maintaining prolonged mTORC1 inhibition when nutrient supplies are critically limited.
Physiological Functions of mTOR Activation
Given the profound geroprotective benefits of rapamycin, aging is increasingly framed through the lens of the "hyperfunction theory," which posits that aging is not a result of passive molecular wear-and-tear, but rather stems from the chronic overactivation of nutrient-sensing pathways that drive cellular hypertrophy, senescence, and sterile inflammation long after developmental growth has ceased 7.
However, the complete or generalized suppression of mTOR signaling introduces significant physiological liabilities, underscoring the pathway's dual role. The activation of mTORC1 is an absolute biological requirement for skeletal muscle maintenance and adaptation. In response to mechanical load (resistance training) and changes in amino acid availability (specifically leucine), localized mTORC1 signaling drives the protein synthesis required for the enlargement of individual muscle fibers (hypertrophy) 1729. Consequently, the chronic, systemic suppression of mTORC1 carries the inherent risk of exacerbating sarcopenia - the age-related loss of skeletal muscle mass and strength that severely impairs functional independence in older adults 17.
Furthermore, the transient activation of the PI3K-Akt-mTOR signaling axis represents an integral component of tissue regeneration. Genetically defined mouse models have demonstrated that robust mTOR activation is required to stimulate epithelial cell proliferation and migration during cutaneous wound healing 527. The application of pharmacological mTOR inhibitors in acute trauma settings actively delays wound closure and impairs tissue regeneration 27. mTOR signaling is also intrinsically tied to the activation and rapid clonal expansion of various immune cells responding to pathogens. Therefore, while dampening mTOR hyperfunction is critical for delaying age-related decline, the persistent, chronic suppression of the pathway can impair systemic immune surveillance, muscle mass preservation, and wound repair 1028.
Mechanisms of Rapamycin-Induced Insulin Resistance
The most significant clinical barrier to the continuous, widespread use of high-dose rapamycin as an anti-aging therapeutic in healthy humans is its well-documented propensity to induce profound metabolic side effects, specifically glucose intolerance, hyperlipidemia, and clinical insulin resistance 2930. Paradoxically, while caloric restriction naturally extends lifespan and concurrently improves insulin sensitivity, continuous rapamycin therapy frequently disrupts systemic glucose homeostasis 2931.
This paradox is driven entirely by the collateral disruption of mTORC2 3032. As previously established, pre-assembled mTORC2 complexes are acutely insensitive to rapamycin. However, continuous, chronic exposure to the drug binds the entire available pool of intracellular FKBP12 and free mTOR monomers. Over time (typically 24 to 48 hours in vitro or several days in vivo), this blockade prevents the generation of new mTOR complexes 181932. As the existing mTORC2 complexes undergo natural proteasomal degradation, they are not replaced, leading to a profound depletion of mTORC2 signaling capacity across highly metabolically active tissues, including skeletal muscle, white adipose tissue, and the liver 183031.
The physiological pathogenesis of rapamycin-induced insulin resistance follows a precise molecular sequence: 1. Loss of Akt Phosphorylation: Functional mTORC2 is the requisite kinase responsible for phosphorylating Akt at its hydrophobic motif (Serine 473) 1931. The disruption of mTORC2 eliminates this crucial activation step, rendering Akt functionally deficient 29. 2. Failure of Hepatic Gluconeogenesis Suppression: In a healthy metabolic state, the binding of insulin to the hepatic insulin receptor activates Akt, which subsequently acts to suppress the expression of key hepatic gluconeogenic genes, specifically Phosphoenolpyruvate carboxykinase (PEPCK) and Glucose 6-Phosphatase (G6Pase) 31. 3. Hepatic Insulin Resistance: The inability to phosphorylate Akt at S473 causes the liver to become completely refractory to insulin signals. The unsuppressed gluconeogenic machinery drives continuous, unregulated hepatic glucose production, resulting in overt systemic hyperglycemia and compensatory hyperinsulinemia 6293031. 4. GSK3 Beta Activation: In skeletal muscle, reduced Akt activity leads to a failure in the inhibitory phosphorylation of Glycogen Synthase Kinase 3 beta (GSK3-beta) at Serine 9 33. Hyperactive GSK3-beta further alters local glucose and lipid metabolism, cementing peripheral insulin resistance 33.
Crucially, experimental models have demonstrated that the geroprotective properties of mTOR inhibition can be functionally uncoupled from its diabetogenic side effects. Female mice engineered to be heterozygous for both the mTOR gene and the mLST8 gene exhibit significantly decreased mTORC1 activity and extended lifespans, yet they maintain completely normal glucose tolerance and insulin sensitivity 2931. This indicates that lifespan extension relies primarily on the targeted inhibition of mTORC1, whereas insulin resistance is an off-target penalty resulting strictly from the loss of mTORC2 182931.
Mammalian Lifespan and Healthspan Extension
Rapamycin remains the most rigorously validated pharmacological intervention for mammalian lifespan extension discovered to date. Preclinical murine models consistently demonstrate that rapamycin administration, even when initiated in mid-to-late life (equivalent to roughly 60 human years), can extend average and maximal lifespan by approximately 10% to 30%, depending on sex, strain, and dosage 79.
The geroprotective profile of rapamycin closely mirrors that of severe dietary restriction. A comprehensive meta-analysis evaluating 167 lifespan studies across eight distinct vertebrate species confirmed that rapamycin extends lifespan to nearly the exact same magnitude as caloric restriction. Notably, this study revealed that other purported anti-aging pharmaceuticals, such as the Type 2 diabetes medication metformin, failed to show clear, universal longevity benefits across the assessed species 34.
Multi-Pathway Intervention and Trametinib Synergy
While isolated mTOR inhibition successfully delays multiple aspects of aging, the current scientific paradigm is shifting toward combinatorial pharmacology aimed at targeting distinct, parallel hallmarks of aging simultaneously 7. Research published in 2025 in Nature Aging demonstrated that combining rapamycin with trametinib yields highly synergistic geroprotective effects 739.
Trametinib is an FDA-approved drug originally developed for oncology that specifically inhibits MEK within the Ras-MEK-ERK (MAPK) signaling pathway. While rapamycin monotherapy dampens anabolic hyperfunction and reactivates autophagy via mTORC1, trametinib independently reduces inflammaging. It achieves this by dampening pro-inflammatory cytokine amplification (significantly reducing the expression of chemokines such as Cxcl11, Ccl8, and Cd5l) and reducing microglial and astrocyte activation in the brain 7.
When tested independently in middle-aged mice, researchers found that trametinib administration conferred an average lifespan extension of 7.2% in females and 10.2% in males, while rapamycin monotherapy provided an extension of 17.4% and 16.6%, respectively 739. However, when combined, the dual-pathway approach resulted in a striking average lifespan extension of 34.9% in female mice and 27.4% in male mice 739.
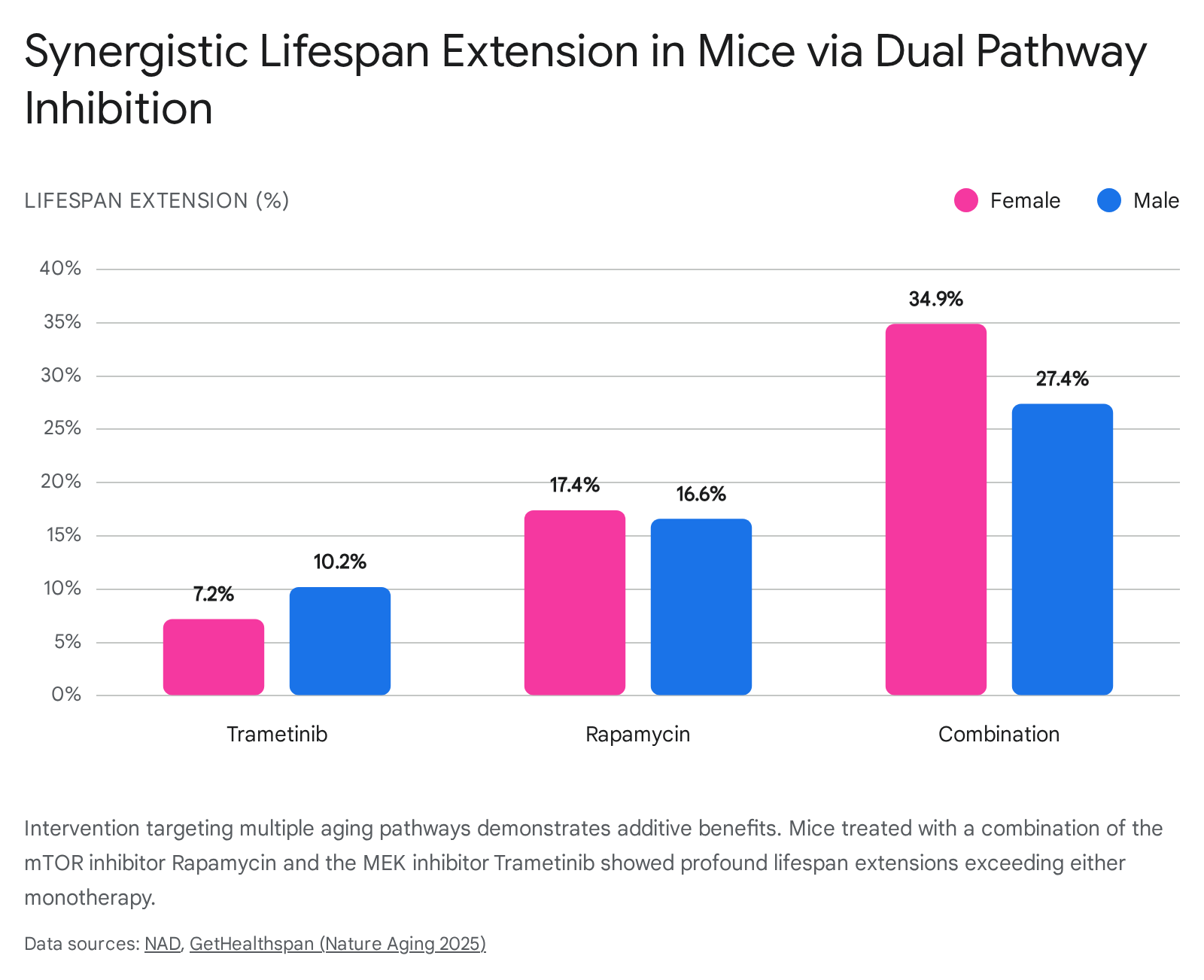
This outcome significantly exceeds the lifespan extension observed when either drug is administered in isolation, supporting the necessity of a systems-level strategy to modulate interrelated aging networks concurrently 7.
Direct Genoprotection and Cellular Senescence
Beyond metabolic restructuring, recent investigations indicate that mTOR inhibition directly addresses genomic instability. Cellular senescence occurs when highly damaged cells permanently exit the cell cycle, secreting a cocktail of molecules that promote chronic low-grade inflammation (the Senescence-Associated Secretory Phenotype, or SASP) 81016.
Research evaluating primary human T cells demonstrated that rapamycin prevents cell death and senescence resulting directly from chemically induced DNA damage. When T cells were exposed to the DNA-damaging antibiotic zeocin, rapamycin administration increased the cell survival rate by three-fold 816. Furthermore, ex vivo analysis of aged immune cells from healthy older adults confirmed that populations with hyperactivated mTORC1 exhibited a stark enrichment for senescence markers. Following targeted rapamycin treatment, expression of p21 - a primary marker of DNA damage-induced senescence - was significantly reduced 8. Rapamycin thereby acts as a potent senomorphic agent, limiting the acquisition of a pro-inflammatory phenotype and enhancing systemic resilience against accumulating genomic instability 81016.
Clinical Applications and Dosing Paradigms
Translating rapamycin's robust preclinical success into practical human therapies requires navigating a highly complex pharmacological landscape. The fundamental challenge lies in harnessing the longevity-promoting benefits of mTORC1 inhibition - such as restored autophagy and proteostasis - while actively avoiding the metabolic and immunosuppressive toxicities associated with mTORC2 suppression 3035.
The PEARL Clinical Trial Outcomes
To address the severe paucity of rigorous clinical data regarding off-label rapamycin use in healthy aging humans, the Participatory Evaluation of Aging with Rapamycin for Longevity (PEARL) trial was conducted and published in late 2024 3642. This decentralized, 48-week, double-blind, randomized, placebo-controlled trial evaluated healthy older adults taking either 5 mg or 10 mg of compounded rapamycin per week 42. A total of 114 participants completed the exhaustive 48-week protocol 37.
The primary safety outcomes demonstrated that intermittent, low-dose rapamycin administration was well-tolerated over the year-long period, with no significant differences in moderate-to-severe adverse events or blood safety biomarkers (such as alarming alterations in lipids, liver enzymes, or glucose metabolism) compared to the placebo cohort 4237.
Efficacy outcomes, evaluated via extensive DEXA scans and standardized surveys, were highly sex-specific. Women in the 10 mg weekly treatment group experienced the most robust, statistically significant improvements, noting a 4.5% increase in lean tissue mass, alongside significant reductions in self-reported pain (via WOMAC indices) and improvements in general health and social functioning 363744. Conversely, men in the 10 mg weekly cohort demonstrated a significant increase of 1.4% in bone mineral content (BMC) 3642.
It is critically important to note a major limitation uncovered during the PEARL trial: pharmacokinetic analysis revealed that the compounded rapamycin utilized for the study was approximately 3.5 times less bioavailable than commercially manufactured formulations 3637. Consequently, the 5 mg and 10 mg cohorts received an effective systemic dose equivalent to roughly 1.4 mg and 2.9 mg, respectively 36. While this implies that the observed benefits occurred at functionally very low doses, it also complicates the direct translation of these dosages to clinical practice utilizing highly bioavailable commercial tablets. Furthermore, while the trial showed functional improvements, researchers caution that there remains insufficient clinical evidence to definitively affirm or negate whether rapamycin directly extends human lifespan, necessitating vastly larger, multi-decade longitudinal studies 45.
Continuous versus Intermittent Dosing Paradigms
The clinical viability of rapamycin in longevity medicine hinges strictly on the architecture of the dosing schedule. The conventional clinical application of rapamycin for solid organ transplant recipients relies on continuous, daily dosing aimed at maintaining high systemic trough concentrations 1046. This protocol is explicitly designed to induce profound immunosuppression, but it frequently results in the severe depletion of mTORC2, precipitating dyslipidemia, delayed wound healing, and drug-induced diabetes 91046.
For longevity and healthspan optimization, the field has universally pivoted toward low-dose intermittent paradigms (typically 3 to 10 mg administered once weekly or bi-weekly) 354647.
| Pharmacological Implication | Continuous / Daily Dosing (Transplant Protocol) | Intermittent / Weekly Dosing (Longevity Protocol) |
|---|---|---|
| mTORC1 Inhibition | Persistent, profound suppression | Pulsatile, transient suppression |
| mTORC2 Inhibition | Inhibited (due to complex assembly blockade) | Preserved (allows complex rebound between doses) |
| Metabolic Health | Promotes insulin resistance, hyperglycemia, hyperlipidemia | Maintains glucose tolerance and lipid homeostasis |
| Immune System Effects | Potent immunosuppression, increased infection risk | Immunomodulatory, enhanced vaccination response |
| Cellular State | Suppresses tissue repair, inhibits muscle hypertrophy | Promotes cyclic autophagy while allowing episodic anabolism |
By providing a substantial temporal gap between drug exposures, intermittent protocols exploit the differing pharmacokinetics and structural sensitivities of the two mTOR complexes. mTORC1 is acutely sensitive to the rapid spike in rapamycin concentration following an oral dose, effectively halting mRNA translation and triggering cellular maintenance processes like autophagy 35. However, as the drug clears the system before the next weekly dose, mTORC1 activity is allowed to naturally rebound, permitting necessary protein synthesis, tissue repair, and immune cell proliferation 46. Crucially, this transient exposure prevents the gradual depletion of free mTOR monomers required for mTORC2 assembly, thereby protecting the patient's glucose regulation and basal insulin sensitivity 3035.
Immunomodulation and the Dosing Threshold
Perhaps the most counterintuitive discovery regarding mTOR inhibition is its distinctly biphasic effect on the immune system. While high, continuous doses are overtly immunosuppressive, low, intermittent doses exhibit potent immunostimulatory and rejuvenating properties 102838.
Landmark clinical trials evaluating the rapalog everolimus (RAD001) in healthy older adults definitively demonstrated this threshold effect. Patients receiving low, intermittent doses (such as 5 mg weekly) exhibited a 20% enhancement in their adaptive immune response to the influenza vaccine 463950. This immune enhancement was accompanied by a significant decrease in the proportion of functionally exhausted, senescent (PD-1 positive) CD4 and CD8 T cells, suggesting a more youthful immune phenotype 4639.
This identifies a critical immunological threshold: below a specific concentration and frequency, mTOR inhibitors alleviate immune exhaustion, rejuvenate hematological stem cell populations, and promote the formation of memory T cells 103950. However, above that threshold, the persistent suppression of mTORC1 overrides these benefits, arresting leukocyte proliferation and inducing broad alloreactive tolerance 102839.
Conclusion
The mechanistic target of rapamycin pathway operates as the central metabolic nexus, evaluating nutrient availability to govern the fundamental pace of biological aging. While the unconstrained hyperfunction of mTORC1 drives cellular senescence, macromolecular damage, and age-related physiological decline, the precise pharmacological modulation of this pathway via rapamycin represents a profound tool for expanding healthspan.
Extensive preclinical data across multiple vertebrate species, increasingly bolstered by emerging human clinical trials such as PEARL, confirm that rapamycin's utility in geroscience is inextricably linked to its dosing architecture. Intermittent, low-dose administration successfully isolates the geroprotective benefits of mTORC1 inhibition - including enhanced autophagic clearance, restored proteostasis, and profound immune rejuvenation - while largely circumventing the diabetogenic and immunosuppressive penalties caused by the inadvertent disruption of mTORC2.
Furthermore, the future of pharmacological longevity interventions will likely rely on polypharmaceutical approaches, combining mTOR inhibitors with complementary agents like trametinib to target the multifactorial nature of systemic aging synergistically. While long-term human lifespan outcomes remain decades away from definitive clinical validation, the strategic, pulsatile modulation of the mTOR signaling network currently stands as the most scientifically substantiated and biologically plausible paradigm in longevity medicine.