Proteostasis decline in aging and neurodegeneration
Evolution and Conceptual Framework of the Proteostasis Network
Cellular protein homeostasis, universally recognized in contemporary molecular biology as proteostasis, encompasses the elaborate and highly integrated network of biological pathways that govern the synthesis, folding, trafficking, and degradation of the cellular proteome. The conceptual framework of the proteostasis network was formally introduced and synthesized in 2008 through the collaborative efforts of biologists and chemists, including Balch, Morimoto, Dillin, and Kelly 1234. Their foundational work established that proteostasis is not merely a collection of isolated quality control mechanisms, but rather an interconnected, dynamic biological system adapted to manage the proteome in response to physiological demands, developmental cues, and environmental stressors 356. Research over the subsequent two decades has rigorously defined this network as an evolutionarily conserved functional unit comprising approximately 1,000 to 1,400 distinct genetic components in human biology, extending beyond foundational protein synthesis to include highly specialized molecular chaperones, intricate degradation machineries, and compartment-specific stress-response signaling pathways 17.
The evolutionary origins of the proteostasis network reveal that it has co-evolved closely with the proteome to regulate the physiological state of the cell across all domains of life, reflecting the unique stresses that different cellular environments experience. In Bacteria and Archaea, the network relies on ATP-driven AAA+ proteases and primary chaperonins, but the division of proteostatic labor was markedly elaborated in Eukarya 18. Eukaryotic cells developed multiple subcellular compartments with specialized functions to facilitate folding biology, including membrane trafficking organelles, the endoplasmic reticulum, and energy-producing compartments like mitochondria, each requiring distinct, localized proteostatic sensors and effectors 19. This evolutionary expansion fundamentally links phenotype and genotype, as the network closely manages the capacity of organisms to tolerate genetic variation and environmental variation without succumbing to proteotoxicity 18.
To conceptualize precisely how proteins fold and function within the constraints of this crowded cellular environment, researchers developed the thermodynamic "FoldFx" model. The FoldFx model integrates the inherent chemical, thermodynamic, and kinetic properties of a polypeptide chain with the real-time capacity of the cellular proteostasis network 41011. A central tenet of the FoldFx model is the definition of the "proteostasis boundary," an abstract mathematical and conceptual threshold within a three-dimensional energetic space. This space is defined by folding thermodynamics (from unstable to stable), folding kinetics (from slow to fast), and misfolding kinetics (from slow to fast) 510.
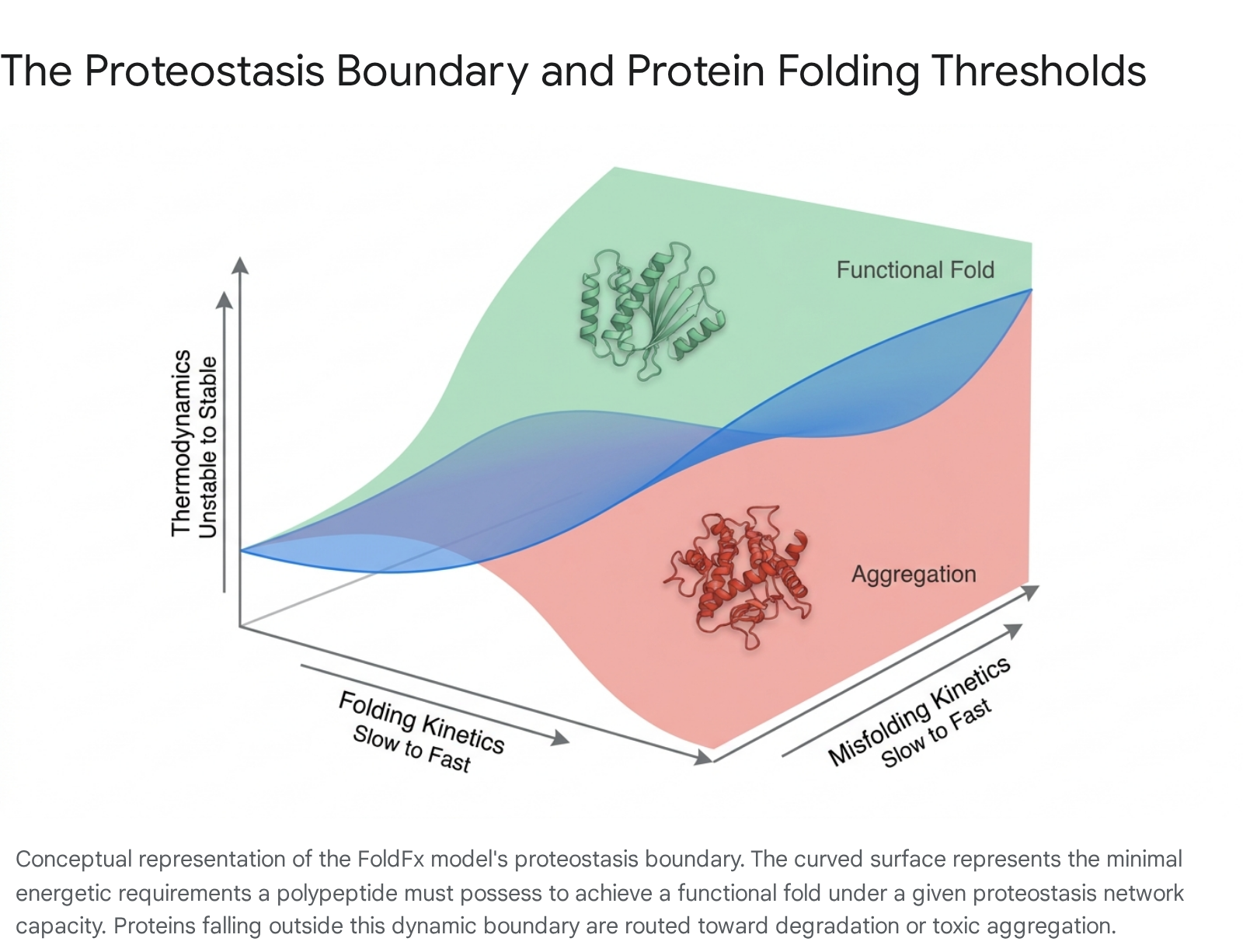
The proteostasis boundary dictates whether a given protein will achieve its mature functional conformation, or whether its energetic properties will cause it to succumb to aberrant misfolding and subsequent aggregation 41213.
The capacity of this theoretical boundary is not fixed across the lifespan of the organism; it is continuously and dynamically adjusted by compartment-specific stress responses. Notable examples include the cytosolic heat shock response, the unfolded protein response of the endoplasmic reticulum, and the mitochondrial unfolded protein response 91014. By upregulating specific subsets of chaperones and targeted clearance factors, these signaling cascades effectively expand the proteostasis boundary to accommodate transient increases in the cellular folding load, or to safely manage the accumulation of misfolded intermediates 101515. However, when intrinsic genetic mutations, environmental toxins, or the systemic biological processes of aging push the cellular proteome definitively beyond the compensatory limits of the network, a widespread proteostasis collapse occurs. This collapse precipitates the accumulation of highly proteotoxic aggregates that directly drive the pathogenesis of severe neurodegenerative diseases 131617.
Core Mechanisms of Cellular Protein Quality Control
Maintaining the structural integrity of the proteome relies on a tripartite system of coordinated quality control. This system involves molecular chaperones that mediate initial folding and conformational maintenance, the ubiquitin-proteasome system designed for the targeted degradation of short-lived proteins, and complex autophagic pathways tasked with the clearance of larger protein aggregates and damaged organelles.
Molecular Chaperones and Conformational Maintenance
Molecular chaperones function as the primary sentinels of the proteostasis network, acting at nearly every stage of a protein's lifecycle. An estimated 280 distinct components participate directly in nascent chain folding alone 7. These factors ensure de novo folding in the incredibly crowded cellular environment, prevent aberrant inter-molecular interactions, and actively direct terminally misfolded proteins toward appropriate degradation pathways 17. The human genome encodes a highly diverse array of chaperones, which are broadly categorized into distinct families based on their molecular weight, domain structure, and specific physiological function. Prominent among these are the HSP40, HSP70, and HSP90 families, as well as the HSP60 family of chaperonins (such as the eukaryotic TRiC/CCT complex), which form specialized cylindrical folding chambers that transiently isolate proteins in productive folding environments 111.
The activity, transcription, and steady-state expression of these cytosolic chaperones are tightly regulated by heat shock transcription factor 1 (HSF1), which serves as the master regulator of the cytosolic heat shock response 1. Under basal, homeostatic conditions, HSF1 is sequestered in an inactive, monomeric state through direct inhibitory binding with HSP90 118. Upon exposure to proteotoxic stress - where denatured or unfolded proteins rapidly accumulate and compete for the HSP90 chaperone pool - HSP90 is titrated away from HSF1. The liberated transcription factor undergoes rapid phosphorylation, forms an active homotrimer, and translocates into the nucleus 1. Once in the nucleus, trimeric HSF1 binds with high affinity to heat shock elements located in the promoter regions of target genes, driving the massive upregulation of over 1,000 stress-response proteins designed to restore homeostasis 118. Importantly, molecular chaperones do not merely fold proteins passively; they actively dictate the ultimate fate of misfolded substrates by interacting with a suite of highly specific co-chaperones. Co-chaperones containing tetratricopeptide repeat domains (such as CHIP and HOP) or BAG domains (such as BAG1 through BAG6) function as critical adapters that physically route aberrant substrates to either the proteasome or the lysosome for terminal destruction 1119.
The Ubiquitin-Proteasome System
For proteins that fundamentally fail to reach their native functional conformation, or those that have successfully completed their intended functional lifespan, the ubiquitin-proteasome system serves as the primary and most highly trafficked route for targeted degradation. The ubiquitin-proteasome system is an ATP-dependent, non-lysosomal proteolytic mechanism responsible for degrading the vast majority (approximately 80% to 90%) of all intracellular proteins 1520. It is particularly specialized for managing short-lived, soluble, or moderately misfolded proteins situated within the cytosol and the nucleus.
Degradation via this pathway is initiated by a highly specific, sequential enzymatic cascade involving ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3). This cascade covalently attaches a polyubiquitin chain to the target substrate, utilizing the hydrolysis of ATP to drive the reaction 151521. The exact structural topology of the resulting ubiquitin linkages dictates the substrate's ultimate cellular fate. Specifically, ubiquitin chains linked via lysine-48 (K48) serve as the canonical recognition signal for immediate proteasomal destruction, whereas lysine-63 (K63) linkages are more frequently associated with signal transduction, DNA repair, or targeting toward autophagic pathways 152122.
The K48-ubiquitinated substrate is subsequently recognized by the 19S regulatory caps of the massive 26S proteasome complex. Specialized subunits within the 19S cap, notably Rpn10 and Rpn13, bind the ubiquitin chain, while the intrinsic ATPase subunits of the complex actively unfold the protein and thread it linearly into the barrel-shaped 20S core particle 2324. Within the sequestered environment of the 20S core, active site threonine residues proteolytically cleave the substrate into short, harmless peptides that are released back into the cytosol 23. When the sheer capacity of the ubiquitin-proteasome system is overwhelmed - either by a massive stress-induced influx of misfolded proteins or by direct physical inhibition from intractable aggregates - the cell attempts to mitigate toxicity by actively sequestering the excess load. Utilizing an intact microtubular network, the cell retrogradely transports these ubiquitinated proteins to the microtubule organizing center, forming specialized, juxta-nuclear inclusion bodies known as aggresomes. These structures serve as temporary holding sites, routing the aggregated proteins away from the stalled proteasome and toward eventual clearance via autophagy 202225.
Lysosomal and Autophagic Degradation Pathways
While the ubiquitin-proteasome system is highly efficient at degrading individual, soluble polypeptides, it is structurally incapable of processing large, bulky aggregates. The autophagy-lysosome system is essential for the clearance of these long-lived proteins, massive macromolecular complexes, and whole dysfunctional organelles 202627. Autophagic processes are highly diverse in their mechanisms of substrate recognition and delivery, and can be broadly categorized into distinct pathways to manage different classes of cellular debris.
| Autophagic Pathway | Mechanism of Substrate Delivery | Primary Substrates | Key Molecular Mediators |
|---|---|---|---|
| Macroautophagy | Sequestration of cytoplasmic regions within de novo double-membrane autophagosomes, followed by ultimate fusion with lysosomes. | Whole organelles (mitochondria), bulk cytoplasm, large protein inclusions, and intracellular pathogens. | ULK1 complex, LC3, specific cargo receptors (p62/SQSTM1, OPTN, NBR1, TAXBP1). 152728 |
| Chaperone-Mediated Autophagy (CMA) | Direct, vesicle-independent unfolding and translocation of individual proteins directly across the lysosomal membrane. | Soluble cytosolic proteins containing a highly specific KFERQ-like pentapeptide motif (~30% of the proteome). | HSPA8 (HSC70), LAMP-2A (the primary rate-limiting lysosomal receptor). 282930 |
| Chaperone-Assisted Selective Autophagy (CASA) | Chaperone-recognized misfolded proteins are selectively ubiquitinated, enveloped by nascent autophagosomes, and delivered to lysosomes. | Dysfunctional chaperone-bound proteins, structurally strained cytoskeletal proteins (e.g., filamin), Z-disk components. | HSPA8, HSPB8, BAG3, STUB1 (the associated E3 ubiquitin ligase), SQSTM1, SYNPO2. 222527 |
Chaperone-mediated autophagy provides highly selective, molecule-by-molecule degradation of specific targets, functioning entirely independent of vesicle formation. Cytosolic proteins bearing a KFERQ-like sequence motif are directly recognized by the constitutively active cytosolic chaperone HSPA8 (also known historically as HSC70) 272930. The chaperone-substrate complex navigates to the lysosomal surface, where it binds to the cytosolic tail of the Lysosome-Associated Membrane Protein type 2A (LAMP-2A) receptor 2930. The binding induces multimerization of LAMP-2A, forming a translocation channel. The substrate is subsequently unfolded and translocated directly into the lysosomal lumen for rapid degradation by resident acid hydrolases and cathepsins 2729.
Conversely, Chaperone-Assisted Selective Autophagy bridges the highly specific chaperone recognition systems with the bulk-clearance machinery of macroautophagy. In this pathway, complex, multimeric chaperone assemblies - comprising the core chaperones HSPA8 and HSPB8, the vital co-chaperone BAG3, and the E3 ubiquitin ligase STUB1 - recognize and immediately ubiquitinate highly aggregation-prone proteins or mechanically damaged structural proteins 2527. The autophagic adaptor protein p62/SQSTM1 then physically tethers these ubiquitinated complexes to nascent autophagosome membranes 25. Autophagosome formation during this process relies heavily on the interaction between BAG3 and SYNPO2, which triggers downstream fusion events 2225. The intricate interplay between the ubiquitin-proteasome system and these highly specific autophagic pathways ensures a robust, interconnected defense against proteotoxicity, allowing one system to dynamically compensate for the transient failure of another under acute stress conditions 152627.
Age-Related Disruption of Proteostasis
The progressive deterioration of the proteostasis network is universally recognized by the scientific community as a primary, foundational hallmark of biological aging 1416. Rather than a uniform or simple systemic decline, age-related proteostasis collapse manifests as a highly complex, interconnected dysfunction characterized by the deterioration of specific chaperones, profound proteolytic impairment across both the proteasome and lysosome, and severe metabolic uncoupling.
Systemic and Asynchronous Organ Aging
Recent comprehensive biological profiling demonstrates that aging - and the accompanying decline in homeostatic mechanisms - does not occur smoothly or synchronously across the human body. A landmark 2026 study conducted by the Chinese Aging Biomarker Consortium, which meticulously analyzed the multicentric Chinese Aging Standardized (mCAS) cohort of 2,019 healthy individuals spanning ages 18 to 91, utilized a deep-learning "Digital Aging Twin" framework to track over 240 distinct physiological, clinical, and molecular parameters 3132. By integrating multiple layers of molecular data - including DNA methylation, RNA transcripts, proteins, metabolites, and gut microbiomes - the researchers established a three-tiered system of highly accurate biological aging clocks 3132.
The mCAS data revealed two distinct, non-linear waves of accelerated biological aging occurring generally in the fourth and sixth decades of human life 3233. Crucially, the deep-learning organ-specific clocks demonstrated that distinct organ systems age asynchronously; the liver was found to reach a critical inflection point of biological aging near age 40, whereas the brain's aging trajectory accelerates closer to age 50 3233. Investigating the root causes of this systemic decline, the researchers analyzed plasma proteomics and identified that the age-driven accumulation of specific liver-derived coagulation factors - most notably F13B, alongside F9 and F10 - acts cell-non-autonomously as a direct systemic driver of vascular and endothelial senescence 3132. In controlled models, exposing healthy aortic endothelial cells to these specific coagulation factors directly induced cellular senescence, impaired blood vessel tube formation, and massive inflammation 32. These groundbreaking findings highlight that localized proteostatic and metabolic alterations in one specific organ can propagate degenerative signals systemically through the plasma, heavily influencing the aging trajectories and vulnerability of distal tissues like the brain.
Chaperone Overload and Proteasomal Failure
At the cellular level, as tissues age, the efficiency of the molecular chaperone network wanes significantly. Experimental models indicate that aged cells fundamentally fail to mount a robust heat shock response to new stressors 1418. This specific deficit is frequently linked to broader epigenetic alterations in chromatin state and inhibitory post-translational modifications of HSF1, which drastically reduce the availability of newly synthesized, functional chaperones 1418. This reduced chaperone capacity leads to a dangerous stoichiometric imbalance, termed "chaperone overload." In an aging organism, an increasing burden of oxidatively damaged or translationally stalled proteins drastically outstrips the available pool of functional chaperones, facilitating unchecked protein aggregation and leaving the proteome highly vulnerable 34.
Concurrently, the ubiquitin-proteasome system undergoes marked functional deterioration during the aging process. In the aging brain, and particularly during the early, prodromal stages of neurodegeneration, the highly efficient, ATP-dependent 26S proteasome exhibits a systemic tendency to disassemble into the less effective, non-ATP-dependent 20S core particle, drastically lowering the overall degradative flux of the cell 24. Furthermore, the proteasome itself becomes physically vulnerable to structural inhibition. As chaperones fail to prevent aggregation, toxic protein oligomers can physically occlude the narrow entry pore of the proteasome. This initiates a catastrophic, positive-feedback loop where diminished clearance yields further aggregate accumulation, which in turn causes further direct inhibition of the surviving proteasome complexes 2023.
Dysregulation of Autophagy and Lysosomal Integrity
The cellular autophagic clearance pathways are uniquely susceptible to the aging process, suffering declines that closely mirror the failure of the proteasome. The efficiency of chaperone-mediated autophagy steadily decreases in older tissues, including highly metabolic environments like skeletal muscle and the central nervous system 353637. Mechanistically, extensive research has proven that this decline is not primarily due to the loss of the cytosolic chaperone HSPA8, which remains relatively stable with age 35. Rather, the primary defect lies in the age-related destabilization and depletion of the lysosomal receptor LAMP-2A 35. The prevailing "Aging Model" of chaperone-mediated autophagy decline posits that age-associated shifts in the lipid composition of the lysosomal membrane directly disrupt the multimerization, stability, and recycling of LAMP-2A, severing the critical bottleneck for substrate translocation 3839. The systemic reduction in this specific clearance pathway leads to progressive skeletal muscle myopathy in aged animal models, and allows for the aberrant accumulation of substrates directly implicated in neurodegenerative diseases and severe metabolic disorders 363739.
Lysosomal dysfunction during aging is further exacerbated by the breakdown of lipostasis. Emerging research heavily underscores that the lysosome acts as a central metabolic hub regulating both protein and lipid homeostasis simultaneously. Age-related defects in lysosomal sphingolipid metabolism severely compromise lysosomal membrane integrity, diminishing the organelle's capacity to maintain the acidic pH required to execute the terminal degradation of autophagic cargo 40. Consequently, fully formed autophagosomes accumulate in the cytoplasm, loaded with toxic protein species that cannot be effectively cleared due to a failure in final lysosomal fusion and degradation 2641.
Interplay Between Proteostasis and Metabolic Decline
Proteostasis is inextricably linked to cellular metabolism, mitochondrial function, and nutrient-sensing pathways. Chronic metabolic perturbations, such as the systemic insulin resistance and hyperglycemia frequently observed in aging populations, directly impair proteasomal efficiency. This occurs via the direct glycation of essential proteasomal subunits and the widespread induction of severe oxidative stress, which damages the proteolytic machinery 16. Furthermore, the loss of mitochondrial proteostasis - governed by enzymes like the m-AAA protease and its catalytic subunit AFG3L2 - results in defective calcium handling, diminished respiration, and profound energy deficits that starve the ATP-dependent chaperone and proteasome systems 42.
Insights into the successful maintenance of these interconnected systems have been gleaned from robust longitudinal studies of exceptional human longevity. The Septuagenarian, Octogenarian, Nonagenarian Investigation with Centenarian (SONIC) study in Japan - a nation reaching a historic milestone of nearly 100,000 documented centenarians as of late 2025 - highlights the profound impact of long-term lifestyle and diet on mitigating the biological parameters of aging 434544. Strict adherence to the traditional Japanese dietary pattern (which is notably high in soy products, fish, seaweed, and low in highly processed foods) and cultural practices like hara hachi bu (mindful, natural caloric restriction) are tightly correlated with suppressed chronic inflammation, lower midlife mortality, and a drastically reduced incidence of age-related cognitive decline and dementia 454445. Molecular analyses of these exceptional populations using advanced epigenetic clocks indicate that biological aging proceeds significantly more slowly in distinct tissues; for instance, the cerebellum and retina of centenarians appear to age much more slowly than their chronological age, though highly proliferative compartments like the blood may age faster 46. These clinical and epidemiological observations strongly underscore the "Longevity Model" hypothesis of proteostasis, which suggests that lifelong interventions that downregulate the systemic insulin/IGF-1 signaling axis concurrently enhance systemic chaperone-mediated autophagy, preserving autophagic flux, clearing negative regulators of lifespan, and fundamentally extending human healthspan 39.
Proteostasis Collapse in Specific Neurodegenerative Pathologies
The progressive, unmitigated failure of the proteostasis network is the primary pathogenic driver unifying the vast majority of neurodegenerative disorders. Neurons, characterized as highly arborized, post-mitotic, and exceptionally metabolically active cells, are exquisitely vulnerable to the accumulation of misfolded proteins over a human lifespan 2642. When the localized clearance systems fail, specific aggregation-prone proteins undergo severe conformational changes. These proteins frequently adopt highly stable, toxic β-sheet rich oligomeric structures that can propagate in a prion-like manner throughout the central nervous system, driving the progressive anatomical spread of pathology characteristic of these diseases 474849. Interestingly, recent quantitative proteomics studies utilizing vertebrate models (specifically, the short-lived African turquoise killifish) demonstrate that prion-like protein condensates - such as those formed by the RNA-binding helicase DDX5 - accumulate even during normal, physiological brain aging, suggesting a foundational baseline of protein aggregation that precedes frank neurodegenerative disease 50.
Amyloid Beta and Tau in Alzheimer's Disease
Alzheimer's disease is characterized pathologically by the massive extracellular accumulation of amyloid-beta (Aβ) plaques and the dense intracellular accumulation of neurofibrillary tangles composed of hyperphosphorylated tau protein 4151. In Alzheimer's disease, the failure of both the ubiquitin-proteasome system and macroautophagy creates a highly destructive, synergistic blockade. Extensive evidence indicates that soluble Aβ and tau oligomers physically impair proteasomal function 2641. As the primary proteasome system fails, the neuron attempts to upregulate macroautophagy as a compensatory survival mechanism; however, due to the aforementioned age-related lysosomal deficits, the resulting autophagosomes fail to efficiently fuse with lysosomes 26. This stalled autophagic flux transforms vulnerable neurons into stagnant reservoirs of toxic Aβ and tau intermediates. Furthermore, BACE2, a critical enzyme involved in Aβ processing and generation, relies on both the ubiquitin-proteasome system and autophagy for its routine degradation; the dual failure of these clearance systems leads to wildly dysregulated Aβ generation, fundamentally compounding the amyloid pathology 41.
Alpha-Synuclein and Parkinson's Disease
Parkinson's disease is clinically defined by the profound loss of dopaminergic neurons in the substantia nigra, and pathologically by the presence of Lewy bodies, which are dense intracellular inclusions primarily composed of aggregated α-synuclein protein 51. In Parkinson's disease, the proteostasis network is compromised at multiple nodes. Crucially, mutant and aggregated forms of α-synuclein severely disrupt chaperone-mediated autophagy. The aberrant α-synuclein binds with abnormally high affinity to the lysosomal LAMP-2A receptor but fails to translocate into the lumen, thereby physically clogging the receptor and completely inhibiting the degradation of other essential cellular proteins that rely on this pathway 2939. Additionally, Parkinson's disease is heavily characterized by the breakdown of localized mitochondrial proteostasis and specific mitophagy. Genetic mutations in the E3 ubiquitin ligase Parkin and the kinase PINK1 fundamentally impair the targeted ubiquitination of damaged mitochondria, leading to a profound cellular energy deficit that further starves the ATP-dependent proteasome and chaperone machineries, ensuring their failure 52.
Polyglutamine Expansion in Huntington's Disease
Huntington's disease is a devastating, incurable autosomal dominant disorder caused by a CAG trinucleotide repeat expansion in the HTT gene, resulting in a mutant huntingtin protein possessing an abnormally long polyglutamine (polyQ) tract 2153. The expanded mutant huntingtin exerts a massive toxic gain-of-function by directly sequestering key components of the molecular chaperone network and the proteasome, thereby crippling the global folding capacity of the entire cell and leaving other proteins vulnerable to misfolding 1921. Complicating the pathology significantly, the wild-type huntingtin protein normally acts as a critical scaffolding protein directly required for the regulation of selective autophagy 19. Therefore, the genetic mutation not only produces a highly aggregation-prone, toxic substrate but also fundamentally dismantles the very autophagic machinery required by the cell to clear it, leading to an incredibly rapid and profound collapse of the neuronal proteostasis network 19.
TDP-43 and Endoplasmic Reticulum Stress in Amyotrophic Lateral Sclerosis
Amyotrophic Lateral Sclerosis (ALS) involves the relentless, selective degeneration of upper and lower motor neurons in the brainstem and spinal cord 5354. While diverse genetic mutations (such as expansions in C9ORF72 or mutations in SOD1) cause familial variants of ALS, the mislocalization, hyperphosphorylation, and aggregation of the RNA-binding protein TDP-43 is observed as a universal pathological hallmark in approximately 90% of both familial and sporadic cases 4953. ALS pathogenesis is strongly linked to severe, early alterations in the folding capacity of the endoplasmic reticulum (manifesting as profound ER stress) and the subsequent failure of Chaperone-Assisted Selective Autophagy 2254. When motor neurons inevitably fail to clear the mounting TDP-43 aggregates via the ubiquitin-proteasome system, their secondary reliance on Chaperone-Assisted Selective Autophagy is often insufficient. This is frequently due to genetic or age-related declines in essential adaptor components like the co-chaperone BAG3 or the autophagic receptor p62/SQSTM1, allowing the toxic aggregates to relentlessly accumulate and drive motor neuron death 224149.
| Disease | Primary Aggregating Protein(s) | Primary Proteostatic Vulnerabilities and Mechanisms of Failure |
|---|---|---|
| Alzheimer's Disease (AD) | Amyloid-β (Aβ), Hyperphosphorylated Tau | Stalled autophagic flux (failure of autophagosome-lysosome fusion); Direct proteasome occlusion by tau oligomers; Dysregulation of BACE2 degradation. 2641 |
| Parkinson's Disease (PD) | α-Synuclein | Blockade of the chaperone-mediated autophagy receptor (LAMP-2A) by mutant α-synuclein; Impaired mitophagy due to PINK1/Parkin kinase and ligase dysfunction. 2952 |
| Huntington's Disease (HD) | PolyQ-expanded Huntingtin (mHTT) | Sequestration and exhaustion of the cytosolic chaperone network; Loss of wild-type HTT's normal function as an essential scaffolding protein for autophagy. 1921 |
| Amyotrophic Lateral Sclerosis (ALS) | TDP-43, SOD1, Dipeptide repeats (C9ORF72) | Severe endoplasmic reticulum stress and Unfolded Protein Response failure; Dysfunction in Chaperone-Assisted Selective Autophagy (CASA) adaptors like BAG3. 4954 |
Emerging Therapeutic Interventions Targeting Proteostasis
As the precise mechanistic links between proteostasis collapse and widespread neurodegeneration have solidified, the global pharmaceutical landscape has dramatically shifted focus. Moving away from highly singular, often unsuccessful, targets, researchers are developing strategies that directly modulate, enhance, or hijack the cell's endogenous protein quality control systems. The 2025 Alzheimer's disease drug development pipeline reflects this massive diversification, currently tracking 138 novel drugs across 182 active clinical trials, representing a profound paradigm shift toward a broader manipulation of foundational metabolic and proteostatic targets 5556.
Targeted Protein Degradation Modalities
Targeted Protein Degradation represents a transformative, paradigm-shifting pharmacological approach that moves the industry beyond traditional target inhibition (known as occupancy-driven pharmacology) to event-driven pharmacology. Targeted Protein Degradation utilizes highly engineered small molecules or biologics to intentionally hijack the cell's endogenous ubiquitin-proteasome system, forcing the catalytic, rapid destruction of disease-causing proteins that were previously considered entirely "undruggable" because they lacked traditional enzymatic active sites 575859.
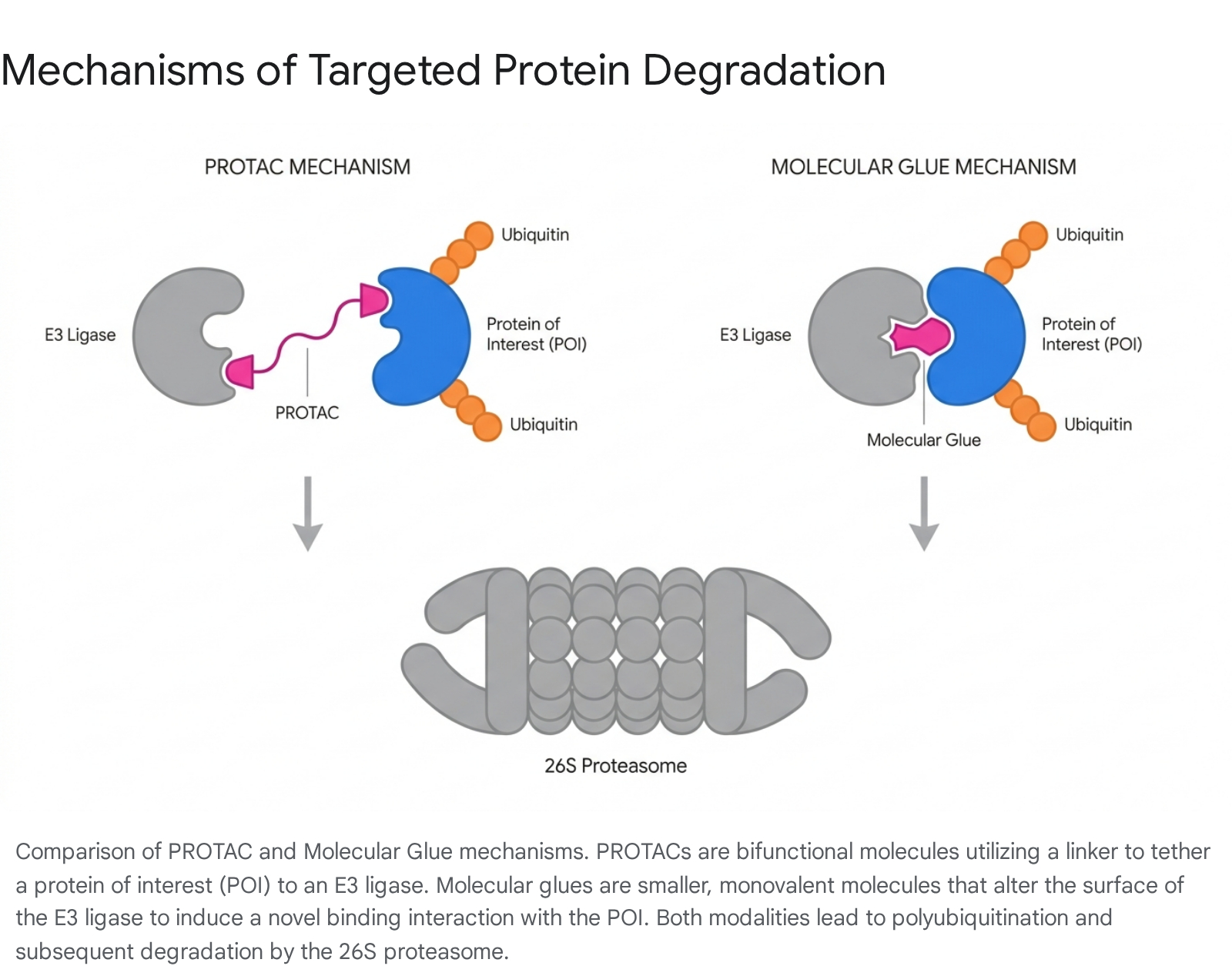
The two primary modalities actively driving the field of Targeted Protein Degradation are Proteolysis-Targeting Chimeras (PROTACs) and Molecular Glues, each with distinct advantages and challenges regarding central nervous system application: * Proteolysis-Targeting Chimeras (PROTACs): These are large, heterobifunctional molecules carefully engineered with three distinct parts: a chemical ligand that selectively binds the pathogenic protein of interest, a separate ligand that recruits a specific E3 ubiquitin ligase (most commonly Cereblon or Von Hippel-Lindau), and a flexible chemical linker connecting the two 5760. By artificially bringing the pathogenic protein and the E3 ligase into close spatial proximity, the PROTAC forces the immediate polyubiquitination and destruction of the target. While highly effective and clinically advanced in oncology, standard small-molecule PROTACs face significant challenges in neurodegenerative applications due to their high molecular weight, which severely restricts blood-brain barrier permeability 5760. However, highly novel biologic approaches are demonstrating profound preclinical success. For example, newly developed bioPROTACs utilizing the E3 ligase CHIP fused to highly specific nanobodies (such as the NbSyn87 nanobody) have proven capable of completely degrading both wild-type and Parkinson's disease-associated mutant α-synuclein in cellular models 61. * Molecular Glues: Unlike the large, complex PROTACs, molecular glues are much smaller, monovalent molecules that completely lack a chemical linker. They function by binding directly to the E3 ubiquitin ligase and slightly altering its three-dimensional surface conformation, thereby creating a novel, high-affinity interface for a neo-substrate that the ligase would not naturally recognize or bind 576263. Classic, FDA-approved examples include the immunomodulatory imide drugs (IMiDs) like thalidomide and lenalidomide, which bind Cereblon to degrade transcription factors in cancers 5763. Due to their significantly smaller size and far more favorable pharmacokinetic profiles, molecular glues hold immense promise for central nervous system penetration. Numerous emerging preclinical compounds are actively being developed to target the degradation of tau, α-synuclein, and even dysregulated signaling kinases via this induced proximity mechanism 525964.
Heat Shock Factor 1 Activation
Rather than focusing exclusively on degrading proteins after they have already misfolded into toxic states, an alternative, preventative strategy aims to preemptively boost the cell's inherent folding capacity before aggregation occurs. Pharmacological activation of the Heat Shock Factor 1 pathway aims to trigger a robust, systemic upregulation of the entire protective chaperone network 1848.
Early experimental drugs such as geldanamycin and its various derivatives function by physically binding to HSP90, disrupting the natural inhibitory complex it forms with HSF1 in the cytosol. This disruption frees HSF1 to immediately migrate to the nucleus and transcribe high levels of protective chaperones like HSP70 1848. While early compounds faced toxicity challenges, newer, highly targeted molecules are showing promise. Compounds demonstrating significant preclinical efficacy include celastrol, which robustly protects dopaminergic neurons in Parkinson's disease models, and oxyphenbutazone, which actively reduces TDP-43 aggregation in ALS cellular models by enhancing HSF1 activity 18. Furthermore, a highly targeted, first-in-class HSF1 pathway inhibitor, known as NXP800, has recently entered Phase I clinical trials in oncology, demonstrating that precise pharmacological modulation of this complex transcription factor network is achievable 65. However, translating HSF1 activators directly to human neurodegenerative diseases requires incredibly careful calibration. Excessive chaperone induction must be avoided, as an overactive proteostasis network is frequently co-opted by oncogenic cells to support rapid, unchecked tumor growth and survival 1865.
Metabolic Repurposing and Synaptic Regeneration
Given the deep, evolutionary integration between proteostasis maintenance and cellular metabolism, repurposing established, safe metabolic drugs has become a major, highly active focus of clinical investigation. Strikingly, approximately 33% of all agents in the 2025 Alzheimer's disease pipeline are repurposed drugs originally approved for other indications 5556. Prominent among these are Glucagon-Like Peptide-1 receptor agonists, such as semaglutide and liraglutide, originally developed and widely used for type-2 diabetes and obesity management 566667. By mitigating systemic insulin resistance, lowering neuroinflammation, and improving energy homeostasis in the brain, these metabolic agents effectively alleviate the extrinsic stress placed on the neuronal proteostasis network. Massive Phase 3 clinical trials evaluating oral semaglutide in early Alzheimer's disease are highly anticipated to yield pivotal, field-altering efficacy data in late 2025 5666.
Concurrently, there is highly active clinical development of completely novel therapies that address the downstream synaptic failure directly caused by prolonged proteostasis collapse. SPG302 (Tazbentetol) is an investigational, once-daily oral agent that acts directly as a synaptic regenerator. Rather than targeting specific protein aggregates, SPG302 targets a critical regulator of the F-actin-based cytoskeleton, actively promoting the de novo growth and stabilization of glutamatergic dendritic spines regardless of the underlying amyloid or tau pathology 686972. In August 2025, a Phase 2a trial of SPG302 in Alzheimer's patients reported remarkable results, demonstrating a nearly 3-point increase in Mini-Mental State Examination scores within just four weeks, with cognitive benefits sustaining through six months of open-label treatment 72. Highlighting its broad potential against neurodegeneration, SPG302 has also received Orphan Drug designation from the FDA and EMA for ALS, where Phase 2a top-line data demonstrated that 82% of treated patients showed a stable or improved rate of clinical decline on the ALSFRS-R scale 6872.
Monoclonal Antibodies and Amyloid Clearance
While expanding the therapeutic focus beyond amyloid is absolutely necessary for comprehensive disease modification, the direct, aggressive clearance of existing toxic aggregates remains a foundational pillar of current Alzheimer's therapy. In July 2025, the European Medicines Agency's Committee for Medicinal Products for Human Use reversed a prior negative opinion, issuing a positive recommendation for the marketing authorization of the advanced anti-amyloid monoclonal antibody donanemab (Kisunla) 707172.
This authorization specifically targets adults with early symptomatic Alzheimer's disease, but importantly, it is restricted to individuals who are apolipoprotein E ε4 (ApoE4) non-carriers or heterozygotes, explicitly excluding ApoE4 homozygotes from the eligible treatment pool 7173. This highly restricted indication stems directly from safety data regarding the incidence of Amyloid-Related Imaging Abnormalities, which occurred at significantly higher rates (55.9%) in patients with two copies of the ApoE4 gene compared to non-carriers (33.0%) 73. This regulatory decision reflects an ongoing, complex effort to balance the definitive clinical benefits of clearing pathogenic aggregates with the localized proteotoxic, vascular, and inflammatory stress such rapid clearance inevitably generates in the brain 7073. To combat these risks and improve efficacy, future paradigms are likely to rely on combination therapies. For instance, combination trials are already emerging, such as the upcoming Phase 3 evaluation of donanemab paired with NA-831, a neuroprotective compound aimed at stimulating neurogenesis while allowing for lower, safer doses of the antibody 74.
Global Consortiums and Future Directions
The sheer complexity of unraveling the proteostasis network and its relationship with systemic aging has necessitated the formation of massive, international research consortiums. Organizations such as the Proteostasis Consortium - bringing together leading researchers from Stanford, Harvard, Northwestern, and UCSF - are systematically annotating the human proteostasis network gene-by-gene and developing open-access tools to identify novel small-molecule regulators of protein folding 7576.
In Europe, major initiatives funded by the European Commission under Horizon 2020 and Horizon Europe are driving the integration of molecular aging markers with population health. The Lifepath consortium has produced extensive data demonstrating how socioeconomic adversity fundamentally accelerates biological aging and chronic inflammation, highlighting that proteostasis collapse is influenced not just by genetics, but by lifelong environmental stressors 7778. Furthermore, newly launched long-term initiatives like the MetAGE project (2024 - 2029) are specifically dedicated to studying the intricate cross-talk between the seven key processes of metabolic control - including lipostasis, mitochondrial function, immunity, and proteostasis - to develop actionable interventions for healthy aging 79. These coordinated, global efforts reflect a unified scientific consensus: effectively treating neurodegeneration requires moving beyond isolated protein targets toward a holistic, systems-level restoration of the organism's foundational protein quality control network.