Programmed aging theories and their scientific rejection
Biological aging represents a progressive decline in physiological integrity, leading to impaired homeostatic regulation, functional degradation, and an exponentially increasing probability of mortality. While the phenotypic manifestations of aging are universally observed across complex mammalian species, the underlying evolutionary and molecular origins of this physiological decline remain the subject of intense, ongoing scientific debate. Within the field of biogerontology, theoretical frameworks are broadly divided into two opposing paradigms: non-programmed (or byproduct) theories, which view aging as the passive accumulation of uncorrected damage or a consequence of evolutionary trade-offs, and programmed theories, which posit that aging is an active, genetically regulated process selected by evolution for specific adaptive advantages 11234.
Despite the intuitive appeal of programmed aging - an appeal frequently bolstered by the discovery of precise biological clocks, highly conserved metabolic pathways, and defined cellular senescence mechanisms - the overwhelming consensus among mainstream gerontologists and evolutionary biologists rejects the notion that aging evolved as a purposeful, organismal termination program. This rejection is rooted deeply in the mathematics of population genetics, the mechanics of individual-level natural selection, and modern computational interpretations of molecular data 5678.
This report provides a comprehensive analysis of programmed aging theories, the molecular mechanisms that purportedly drive them, the alternative quasi-programmed paradigms that bridge theoretical divides, and the foundational evolutionary logic dictating why the biological consensus rejects the premise of adaptive, deliberate organismal death.
Foundational Evolutionary Paradigms of Aging
To analyze the rejection of programmed aging, it is necessary to establish the evolutionary frameworks that form the bedrock of modern biological thought. The debate fundamentally hinges on the level at which natural selection operates and the concept of the evolutionary selection shadow.
The Selection Shadow and Classical Byproduct Theories
The cornerstone of modern evolutionary gerontology, developed in the mid-20th century by J.B.S. Haldane and Peter Medawar, is the concept of the declining force of natural selection with chronological age 5610. In wild populations, animals face constant extrinsic mortality threats from predation, disease, starvation, and environmental hazards. Consequently, very few wild organisms live long enough to experience advanced physiological decay. Because evolution selects for traits based on their ability to increase an individual's reproductive success, genetic mutations that exert harmful effects late in life - after the majority of a cohort has already reproduced or died from extrinsic causes - are essentially invisible to natural selection. This zone of evolutionary blindness is termed the selection shadow 56.
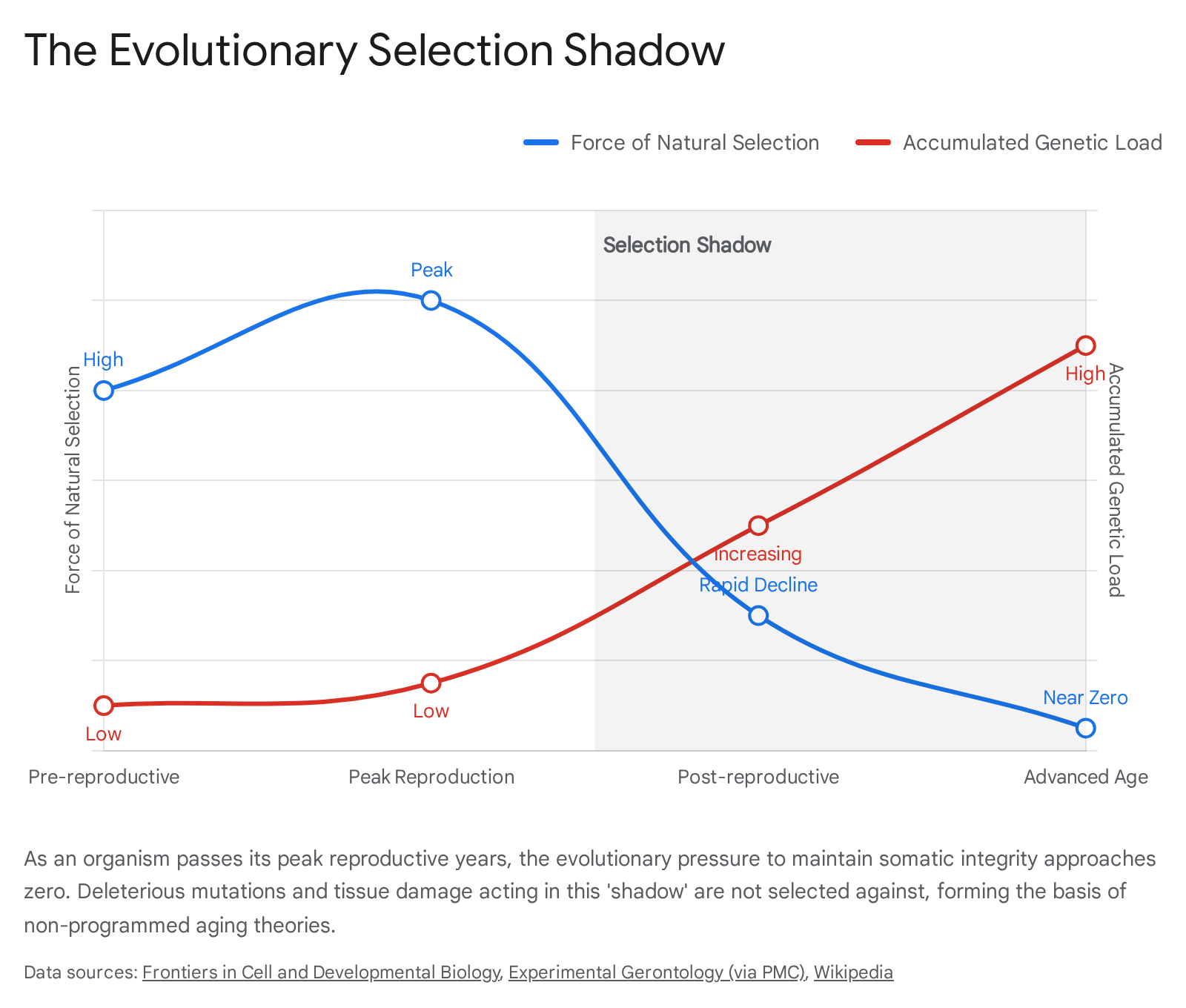
Operating under this premise, three classical byproduct theories of aging emerged. Collectively, these form the non-programmed consensus, asserting that aging is a consequence of evolutionary mechanics rather than a deliberate adaptation.
Mutation Accumulation Theory
Proposed by Peter Medawar in 1952, the Mutation Accumulation (MA) theory suggests that aging arises from the random accumulation of deleterious, late-acting genetic mutations 2356. Because natural selection is too weak to weed out mutations that express themselves only in late adulthood, these mutations passively drift through the population. Over evolutionary time, they create a genetic load that ultimately causes senescence. Under this framework, aging provides no benefit; it is merely an evolutionary oversight 311.
Antagonistic Pleiotropy Theory
Formulated by George C. Williams in 1957, the Antagonistic Pleiotropy (AP) theory posits that certain alleles possess pleiotropic effects, meaning they control multiple, seemingly unrelated phenotypic traits 235711. AP argues that an allele conferring a strong reproductive, metabolic, or developmental advantage early in life will be heavily favored by natural selection, even if that same allele has catastrophic, senescence-inducing effects in post-reproductive life. The evolutionary mathematics dictate that the guarantee of early survival and reproduction massively outweighs the penalty of late-life decay, rendering aging an unavoidable evolutionary trade-off 3611.
Disposable Soma Theory
Proposed by Thomas Kirkwood in 1977, the Disposable Soma (DS) theory frames aging as an optimization problem of metabolic resource allocation 356910. Organisms operate in environments with finite energy resources and must partition these resources between somatic maintenance (e.g., DNA repair, antioxidant defense, proteostasis) and reproduction. Evolution optimizes this allocation to maximize reproductive output, leading to an intentional under-investment in long-term somatic repair 6910. The soma (the physical body) is deemed "disposable" once the germline has been successfully passed to the next generation, resulting in the gradual accumulation of uncorrected cellular damage.
To synthesize the foundational theoretical landscape, Table 1 contrasts these classical byproduct models with programmed and quasi-programmed alternatives.
| Theory Category | Foundational Concept | Evolutionary Rationale | Proposed Mechanism of Action | Key Proponents |
|---|---|---|---|---|
| Byproduct: Mutation Accumulation | Aging as genetic drift | Selection shadow allows late-acting deleterious genes to evade natural selection. | Accumulation of stochastic genetic errors over generations. | Peter Medawar 356 |
| Byproduct: Antagonistic Pleiotropy | Aging as a life-history trade-off | Genes boosting early-life fitness are favored despite late-life harm. | Tumor suppression limits (telomeres); robust immune responses causing late-life inflammation. | George C. Williams 3511 |
| Byproduct: Disposable Soma | Aging as metabolic economics | Energy is prioritized for rapid reproduction over long-term somatic repair. | Under-investment in DNA repair; uncorrected oxidative stress and protein misfolding. | Thomas Kirkwood 3610 |
| Programmed: Phenoptosis | Aging as adaptive suicide | Death clears ecological space for progeny, increasing population evolvability. | Mitochondrial ROS signaling; death hormones; genetically triggered apoptosis. | Vladimir Skulachev 11121314 |
| Quasi-Programmed: Hyperfunction | Aging as a shadow of development | Blind continuation of vital developmental growth programs post-maturity. | Over-activation of mTOR and nutrient-sensing pathways leading to cellular hypertrophy. | Mikhail Blagosklonny, Vladimir Dilman 1151916 |
Evolutionary Objections to Programmed Aging
The primary barrier to the acceptance of programmed aging lies in the mechanics of natural selection. Because aging and death are fundamentally detrimental to the individual organism, proponents of programmed aging must invariably invoke the concept of group selection 17181920.
The Group Selection Debate and the Cheater Problem
Group selection postulates that traits can evolve because they benefit the population, the species, or the ecosystem as a whole, even if they inflict a severe fitness cost on the individual possessing them 171820. Early evolutionary theorists, following the logic of August Weismann, suggested that aging evolved as an altruistic mechanism to clear ecological space and resources for younger, newly adapted generations, thus preventing the stagnation of the species 26111325.
However, the modern evolutionary synthesis, popularized by figures such as W.D. Hamilton, John Maynard Smith, and Richard Dawkins, heavily favors gene-centered and individual-centered selection 182127. Mathematical modeling of group selection reveals a fatal flaw known as the "cheater problem." If a population of organisms evolves an altruistic program to die at a specific age for the good of the species, a single random mutation that disables this aging program (a "cheater" gene) would allow that individual to live longer, consume more resources, and produce vastly more offspring 1827. Within a few generations, the cheater gene would thoroughly dominate the gene pool, and the altruistic aging program would be eradicated.
Multi-Level Selection and Evolvability
In response to the cheater problem, modern advocates for programmed aging utilize multi-level selection models and the concept of "evolvability" 14192021. Theorists argue that while individual selection acts against aging, group-level benefits - such as increased genetic turnover, enhanced ability to adapt to rapid environmental changes, and the stabilization of ecosystem dynamics - exert a simultaneous selective pressure that preserves the aging program 2111425.
Some researchers utilize formalisms like the Price equation to demonstrate that under highly specific demographic conditions, group-level benefits could mathematically counterbalance the individual disadvantage of aging 171921. Despite these models, the mainstream consensus maintains that group selection is inherently weak compared to individual selection, particularly for a trait as drastically fitness-reducing as death. Consequently, the biological community overwhelmingly concludes that a complex, deliberate program for aging lacks a robust Evolutionary Stable Strategy (ESS) 8132127.
Explicit Theories of Programmed Aging
Despite formidable theoretical hurdles, a minority of gerontologists and molecular biologists point to compelling physiological evidence that aging behaves like a highly coordinated, deterministic program. These explicit programmed theories challenge the byproduct consensus by identifying specific mechanisms that appear designed to terminate the organism.
Phenoptosis and the Russian School
The most explicit modern articulation of programmed aging is the concept of "phenoptosis," introduced by the Russian biogerontologist Vladimir Skulachev in 1999 111322. Deriving the term from apoptosis (programmed cell death), Skulachev posits that phenoptosis represents the programmed death of an entire organism, orchestrated actively by its own genome 1122.
Skulachev categorizes phenoptosis into two primary forms: * Acute Phenoptosis: Immediate, rapid death following a specific life event, typically reproduction (known ecologically as semelparity). Classic examples include Pacific salmon, which undergo rapid, hormonally driven degeneration following spawning. In these organisms, aging traits such as massive immunosuppression, organ failure, and the accumulation of amyloid brain plaques appear on the scale of days 111213. Similar acute death programs are observed in marsupial mice and certain cephalopods 1113. In these specific cases, death is indisputably an active, genetically triggered event. * Slow Phenoptosis: The gradual aging process observed in humans and higher mammals. Skulachev argues that slow aging acts to steadily decrease an organism's fitness over time, thereby creating a gradient of vulnerability. This gradient facilitates the natural selection of slightly more fit individuals over less fit older individuals, ultimately accelerating the evolutionary progress of the species 111213.
The primary molecular mechanism proposed for slow phenoptosis involves mitochondria-generated reactive oxygen species (ROS). Rather than viewing ROS strictly as accidental metabolic byproducts causing stochastic damage, Skulachev's framework positions them as deliberate signaling tools utilized by the cell to stimulate apoptosis and steadily degrade tissue cellularity over the lifespan 21222. Operating on this premise, Skulachev directed the SkQ Megaproject, synthesizing SkQ-type substances (plastoquinonyl decyltriphenylphosphonium) designed to penetrate the negatively charged interior of mitochondria and neutralize ROS at the source 111222. The project theorized that this would interrupt the phenoptotic death program. Clinical trials of highly diluted SkQ1 in Russia have resulted in approved treatments for dry eye syndrome and demonstrated lifespan extensions in specific models of fungi, invertebrates, and mammals 1222.
Mainstream gerontologists, however, largely reject the extrapolation of phenoptosis to higher mammals. The consensus classifies the acute death observed in salmon not as aging, but as a highly specialized, stress-induced reproductive strategy - an extreme manifestation of resource reallocation under the Disposable Soma framework - that cannot be analogized to the gradual decline of iteroparous species like humans 1323.
The Information Theory of Aging (ITOA)
Recently, David A. Sinclair and colleagues advanced the Information Theory of Aging (ITOA), which posits that aging is driven not by irreversible sequence mutations in the DNA blueprint, but by the progressive loss of youthful epigenetic information 30242533. In biological systems, genetic information is digital and robust, whereas epigenetic information (such as DNA methylation and histone modifications) is analog and highly susceptible to environmental noise, damage, and metabolic stress 2426.
According to ITOA, cells lose their functional identity as the epigenome becomes increasingly disorganized over time. Crucially, the theory asserts that mammalian cells retain a "backup copy" of their youthful epigenetic state. By utilizing epigenetic reprogramming - specifically, the transient expression of Yamanaka factors (Oct4, Sox2, Klf4) - researchers have demonstrated the ability to theoretically restore this lost information, successfully reverting aged cells to youthful states and restoring vision in aged mouse models 30253536.
To substantiate the claim that epigenetic scrambling directly causes aging, Sinclair's team engineered a mouse model (ICE mice) expressing a specific restriction endonuclease, I-PpoI, which creates controlled double-strand DNA breaks. They argued that the act of faithful DNA repair physically distracts epigenetic maintenance proteins from their normal regulatory locations, causing the epigenome to progressively lose its youthful configuration, thereby actively driving the mice to age faster 253627.
Peer-Reviewed Critiques of ITOA
While highly influential in the public sphere, ITOA has faced intense scrutiny and skepticism from prominent biogerontologists. In a series of formal critiques published in Cell, researchers James A. Timmons and Charles Brenner outlined critical physiological and methodological flaws in the experimental design supporting ITOA 362728.
The primary critique centers on the use of the I-PpoI endonuclease. Brenner points out that I-PpoI is highly genotoxic and inherently cytotoxic. Extensive prior literature demonstrates that targeted I-PpoI expression triggers severe p53-mediated DNA damage responses (DDR) and widespread cell elimination within a month 3627. Critics argue that the accelerated deterioration observed in the ICE mice does not constitute a clean demonstration of epigenetic information loss driving natural aging. Instead, it represents a profound, artificial pathology resembling extreme radiation poisoning or chronic genotoxic stress 36.
Thus, the assertion that ITOA proves aging is an active, reversible epigenetic program remains highly contested. The consensus views the observed epigenetic degradation not as a programmed driver of aging, but as a downstream manifestation of massive, unresolvable stochastic damage overwhelming the cell's repair capacity 15332629.
Biomarkers of Aging and Divergent Interpretations
The debate between programmed and byproduct theories frequently centers on how specific biological biomarkers are interpreted. Molecular features that appear designed to limit lifespan are often reinterpreted by consensus biologists as evolutionary trade-offs.
Telomere Attrition and the Hayflick Limit
Telomeres, the protective nucleoprotein caps located at the ends of linear chromosomes, shorten with each cell division due to the inability of DNA polymerase to fully replicate the lagging strand - a phenomenon known as the "end replication problem" 3031. When telomeres reach a critically short length (the Hayflick limit), cells enter a state of replicative senescence, permanently ceasing division and often secreting inflammatory molecules known as the Senescence-Associated Secretory Phenotype (SASP) 3233343536.
Proponents of programmed aging argue that telomere attrition acts as a deliberate, species-specific molecular countdown clock designed to enforce a finite lifespan and terminate the organism 3547. However, evolutionary biologists counter that telomere shortening evolved via Antagonistic Pleiotropy as a robust tumor-suppressor mechanism 303447. By strictly limiting the number of times a somatic cell can divide, the organism significantly reduces the risk of rampant, uncontrolled cellular proliferation (cancer) during its reproductive peak. The late-life consequences - exhaustion of stem cell pools and the accumulation of toxic senescent tissues - are the acceptable evolutionary costs paid for early-life cancer protection 303436.
Furthermore, telomere shortening is neither universal across species nor the sole cause of telomere dysfunction. Mice, for example, possess telomeres up to ten times longer than humans and widely express telomerase in somatic tissues, yet they live only a fraction of the human lifespan 3137. Additionally, stochastic oxidative stress can cause severe single-strand breaks directly at the telomeric regions, triggering senescence regardless of the telomere's overall base-pair length 3133.
The diversity of progeroid (premature aging) syndromes further complicates the programmed telomere narrative, as outlined in Table 2.
| Progeroid Syndrome | Primary Genetic Defect | Presence of Accelerated Telomere Attrition | Implications for Aging Theory |
|---|---|---|---|
| Werner Syndrome | Mutation in WRN gene (DNA helicase/exonuclease) | Yes, significant attrition observed 3038. | Links specific DNA repair defects directly to telomeric instability and premature senescence 3038. |
| Hutchinson-Gilford Progeria | Mutation in LMNA gene (Lamin A protein) | No consistent attrition universally shared 38. | Indicates severe aging phenotypes can manifest independently of telomere length, driven instead by nuclear envelope instability 38. |
| Dyskeratosis Congenita | Mutations affecting telomerase complex (DKC1, TERC) | Yes, severe attrition is the primary disease driver 38. | Demonstrates the necessity of telomere maintenance for stem cell viability and tissue renewal 38. |
| Wiedemann-Rautenstrauch | Mutation in POLR3A (RNA polymerase III) | No significant telomere shortening identified 38. | Demonstrates that the biological pathways of systemic aging are highly heterogeneous and not solely governed by a single telomeric clock 38. |
This data indicates that telomere attrition is not a universal prerequisite for the manifestation of aging phenotypes, weakening the argument that it serves as a central, programmed death clock 38.
Epigenetic Clocks: Program or Stochastic Drift?
The discovery of epigenetic clocks, pioneered by Steve Horvath, revolutionized quantitative aging research. By measuring DNA methylation states at specific CpG dinucleotide sites, these algorithms can predict the chronological and biological age of an individual with unprecedented accuracy across multiple tissues and mammalian species 73940. The predictability and cross-species conservation of these clocks have reinvigorated claims that aging is a deterministic, genetically scripted program progressing identically across diverse taxa 740.
However, the statistical precision of a clock does not inherently prove the existence of an active aging program. Recent theoretical and computational models have demonstrated that highly accurate biological clocks can emerge purely from stochastic (random) processes.
Research led by Andrew Teschendorff analyzed the mathematical components of epigenetic clocks. The data indicates that clocks which are highly accurate at predicting strictly chronological age are dominated by stochastic DNA methylation drift 41. Conversely, clocks that better predict biological age (healthspan and mortality risk) rely more heavily on non-stochastic, biologically active components 41.
Further challenging the programmed interpretation, researchers David Meyer and Björn Schumacher demonstrated that "aging clocks based on accumulating stochastic variation" can be built using purely simulated, random data 104243. If a biological system begins at a highly ordered ground state (age zero) and accumulates random errors directionally over time, that variation will track chronologically with exceptional precision 1043. Therefore, the existence of highly accurate epigenetic clocks is entirely compatible with non-programmed, wear-and-tear models of aging.
Quasi-Programmed Aging: The Gerontological Middle Ground
The intense polarization between random damage theories (which struggle to explain the highly conserved, predictable pathways of aging) and programmed death theories (which violate foundational evolutionary mechanics) gave rise to a powerful synthesis known as the Quasi-Programmed or Hyperfunction Theory 11519. This framework provides an elegant solution: aging behaves programmatically, but it is not a program for aging.
The Ontogenetic Roots of Elevation Theory
The conceptual origin of quasi-programmed aging rests with Vladimir Dilman, a Soviet endocrinologist who proposed the "Elevation Theory" of aging 16554445. Dilman hypothesized that aging is driven by a progressive, unidirectional loss of sensitivity in the hypothalamus - the brain's master regulator of neuroendocrine homeostasis 554445.
During development, the hypothalamus requires a high threshold of sensitivity to effectively drive the massive metabolic and hormonal changes required for somatic growth and sexual maturation 1655. However, once adulthood is reached, this threshold does not reset. The hypothalamus continues to demand higher levels of peripheral hormones to register a negative feedback loop, forcing the body to chronically elevate hormone and metabolite levels 164445. This state of compensatory metabolic overdrive, termed "age-associated perestroika," ultimately leads to climacteric changes, obesity, hypertension, diabetes, and cancer 5544. Dilman's crucial insight was that age-related pathologies are not the result of systems passively failing, but of regulatory systems permanently stuck in a developmental "push" phase 44.
The Hyperfunction Theory and mTOR
Building directly on his father's legacy, Mikhail Blagosklonny translated Dilman's anatomical hypothesis to the intracellular level, formulating the Hyperfunction Theory of aging 16444558. Blagosklonny identified the mechanistic Target of Rapamycin (mTOR) as the cellular equivalent of Dilman's hypothalamus - a master nutrient-sensing pathway that drives cellular growth, protein synthesis, and proliferation 1151644.
The Hyperfunction Theory operates on a strict, updated interpretation of Antagonistic Pleiotropy. Blagosklonny argues that aging is the "purposeless continuation of developmental growth" 121546. The genetic pathways that build an organism from a single cell into a mature adult cannot simply be switched off once maturity is reached, because there has never been evolutionary pressure to develop a functional post-reproductive "off switch" 47. Consequently, these pathways run on blindly.
Blagosklonny uses the analogy of a "runaway car without brakes" 15. If a car is driving 65 mph on a highway, it is functioning normally. If it continues driving 65 mph into a driveway, it will crash. The crash (aging and disease) is not caused by the rust or wear-and-tear of the car parts; it is caused by the hyper-functional continuation of the driving program in the wrong environment 151958. Under this theory, age-related diseases - such as hypertension, organ hypertrophy, atherosclerosis, and cancer - are not the result of cells failing, but of cells doing too much of what they were designed to do 444748. Cellular senescence is thus recontextualized not as a state of passive exhaustion, but as a state of hypertrophic hyper-activation driven by "geroconversion" 1948.
The Debate Over Primary Damage
The distinction between damage as a primary cause versus a secondary byproduct represents the fundamental theoretical divide in modern biogerontology, highlighted most prominently in scientific exchanges between Mikhail Blagosklonny and Aubrey de Grey.
De Grey, a leading proponent of stochastic damage paradigms, argues that the passage of time and constant metabolic stress lead directly to the accumulation of molecular damage (e.g., DNA mutations, cross-linked proteins, mitochondrial dysfunction). In this framework, this accumulated molecular garbage causes cellular dysfunction, which cascades into organ pathology and ultimately death 1558.
In stark contrast, Blagosklonny's Hyperfunction theory posits that excessive, post-developmental signaling (such as unchecked mTOR activity) directly drives organ pathology through cellular hypertrophy and senescence. Crucially, Blagosklonny asserts that while molecular wear-and-tear certainly exists, it is largely a downstream byproduct of this hyperactive biological state, not the primary, life-limiting cause of death 5848. The organism dies from the pathology of hyperfunction long before it would die from the accumulation of simple molecular rust 1948.
It is critical to emphasize that Blagosklonny explicitly rejected the label of "programmed aging" 11546. A true program possesses an evolutionary intent and a selected outcome. Hyperfunction defines aging as a shadow or a pseudo-program 14648. The growth is purposefully programmed; the resulting late-life destruction is merely an unintended, unselected consequence 46. This framework successfully aligns the predictable, almost deterministic molecular pathways of aging with the rigid strictures of neo-Darwinian evolutionary theory, which forbids the active selection of senescence 94748.
Statistical Predictability and Population Demographics
A lingering argument utilized by programmed aging proponents revolves around the statistical predictability of death. Opponents of programmed aging assert that if aging is merely the accumulation of random, stochastic damage (wear and tear), the variation in the age of death across a population should be enormous, far exceeding the variation seen in genetically scripted developmental milestones.
Research analyzing massive demographic datasets, such as the MIDUS survey in the United States, challenges the assumption that mortality is wildly unpredictable 4950. When comparing the coefficient of variation (relative variability) of strictly programmed developmental events to aging events, the data reveals surprising statistical tightness. The coefficient of variation for the age at menarche (puberty) is roughly 8 - 13%, and for the age at natural menopause it is 7 - 11% 4950. Strikingly, the relative variability for the age at death is 16 - 21% 4950.
While the variation in human mortality is mathematically higher than highly regulated developmental milestones, it is only roughly twice as variable 4950. Programmed aging theorists argue that a purely stochastic, random-damage mechanism should yield vastly wider statistical spreads, suggesting that a loose programmatic clock must be guiding the organism toward a defined biological boundary 4950. However, consensus biologists counter that the inherent structural limits of species-specific anatomical design - coupled with the highly conserved Quasi-Programmed run-on effects of hyperfunction - sufficiently constrain mortality variance without necessitating a genetic suicide program 56.
To summarize the current interpretative divide in geroscience, Table 3 highlights how identical biological observations are interpreted radically differently depending on the theoretical lens applied.
| Biological Biomarker | Programmed Interpretation | Non-Programmed / Quasi-Programmed Interpretation |
|---|---|---|
| Epigenetic Clocks | An active, deterministic software program degrading to enforce lifespan limits 3024. | Stochastic entropy; predictable accumulation of random uncorrected errors moving directionally from a highly ordered ground state 1043. |
| Telomere Shortening | A deliberate molecular countdown timer meant to trigger organismal death 3547. | An imperfect tumor-suppressor mechanism (Antagonistic Pleiotropy) preventing runaway cellular replication in youth 3034. |
| mTOR / Nutrient Pathways | Coordinated metabolic pathways evolved to intentionally shut down somatic maintenance in response to ecological cues 121314. | Blind continuation of vital developmental growth signaling, becoming toxic "hyperfunction" in post-maturity 11547. |
Synthesis and the Current Gerontological Paradigm
The rejection of programmed aging theories by the vast majority of gerontologists is not born of a reluctance to accept complex molecular biology, nor is it a denial of the highly choreographed nature of cellular decline. Rather, it is a strict adherence to the foundational rules of evolutionary mechanics.
Theories advocating for adaptive aging, such as Skulachev's phenoptosis or Sinclair's Information Theory of Aging, rely heavily on group selection - an evolutionary model where individuals sacrifice their own fitness for the demographic health, resource management, or evolvability of the broader species 111320. Because evolutionary success is ultimately defined by individual allele propagation, genetic "cheaters" that ignore the death program would rapidly outcompete their altruistic peers, rendering programmed death mathematically unstable over evolutionary time scales 1827.
The existence of highly predictable epigenetic clocks and conserved metabolic pathways does not override this evolutionary veto. As demonstrated by robust computational modeling, profound predictability can arise purely from the stochastic accumulation of variance beginning from a tightly controlled developmental baseline 104243. Furthermore, what initially appears to be programmed destruction is increasingly recognized as the "shadow" of development. The Quasi-Programmed Hyperfunction Theory effectively bridges the discipline's divide by acknowledging the deterministic, programmatic nature of the cellular pathways driving aging (such as the mTOR complex), while correctly classifying the ensuing destruction as a purposeless, unselected run-on effect governed by Antagonistic Pleiotropy 191547.
Ultimately, the gerontological consensus maintains that aging is the price mammals pay for early-life vitality and reproductive success. It is a highly complex, heterogeneous blend of unselected stochastic damage, unavoidable metabolic resource constraints, and the toxic hyper-activation of vital developmental software that evolution simply never learned to turn off.