Physiology and Pathology of the Blood-Brain Barrier
The blood-brain barrier is a highly selective semipermeable membrane that separates the systemic circulatory system from the central nervous system. Far from a simple passive physical filter, it operates as a dynamic, complex biological interface that strictly controls the influx of essential nutrients, the efflux of metabolic waste, and the exclusion of neurotoxic pathogens and peripheral immune cells. Maintaining the specialized internal microenvironment of the brain is an absolute requirement for proper synaptic transmission, neuronal homeostasis, and cognitive function. Pathological alterations in this barrier are now recognized as critical early events in a wide spectrum of neurological disorders, driving neurodegeneration through both the accumulation of toxic proteins and the initiation of cascading neuroinflammation 123.
Structural and Cellular Architecture of the Neurovascular Unit
The functional integrity of the blood-brain barrier is governed by a multicellular complex known as the neurovascular unit. The concept of the neurovascular unit, developed in the early 2000s, emphasizes that the barrier is not maintained by endothelial cells in isolation, but through continuous, bidirectional communication among several distinct cell types and extracellular matrix components spanning the arteriole-capillary-venule axis 345.
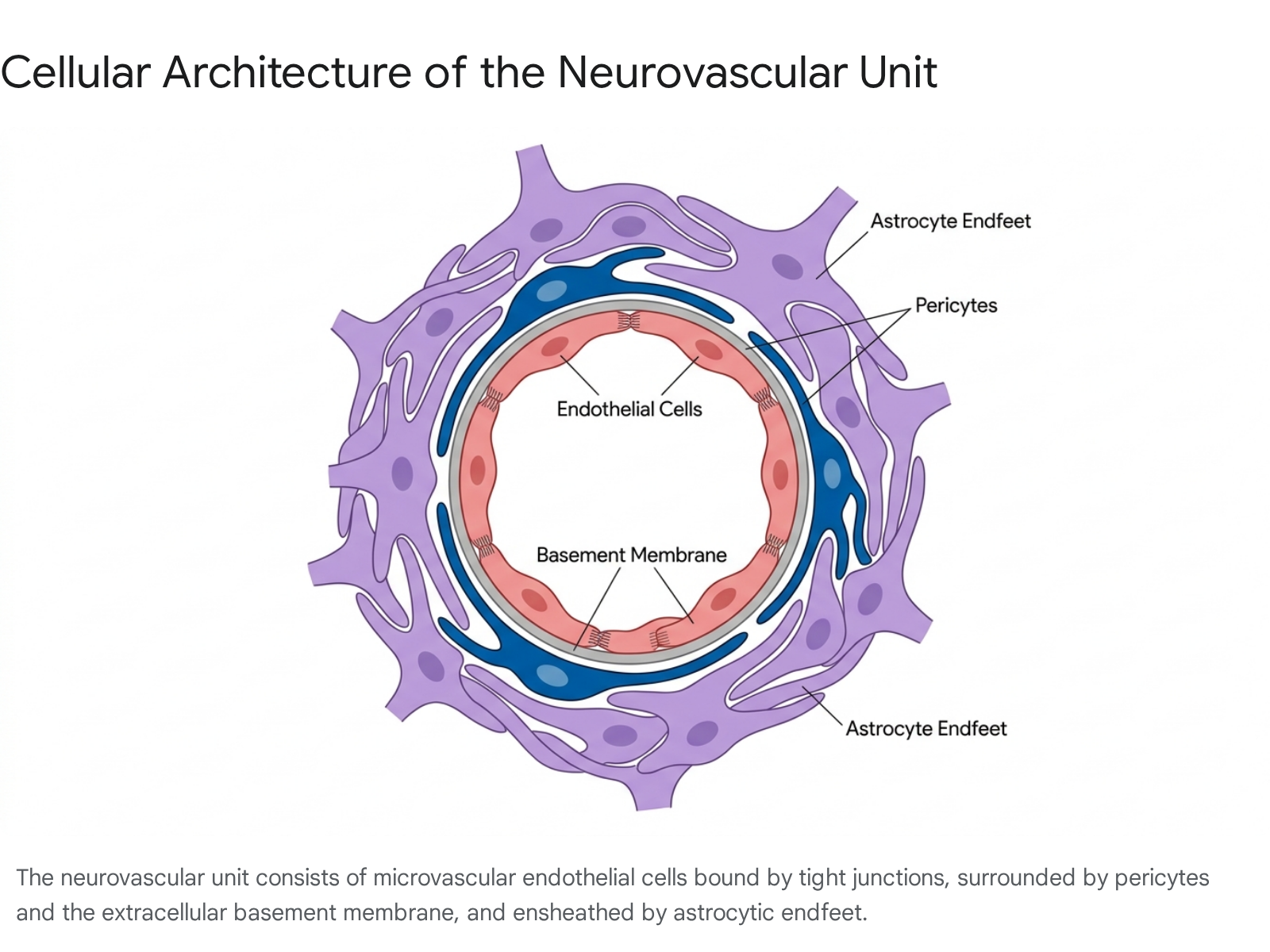
Brain Microvascular Endothelial Cells
At the core of the barrier are brain microvascular endothelial cells, which form the contiguous inner walls of the cerebral capillaries. Unlike endothelial cells in peripheral organs, brain endothelial cells are characterized by a complete lack of fenestrations and extremely low rates of liquid-phase pinocytosis, or transcellular vesicle trafficking 35. This physical morphology effectively eliminates the paracellular diffusion of water-soluble molecules and prevents the bulk flow of blood plasma into the brain parenchyma. Consequently, the transport of required molecules, such as glucose and amino acids, relies entirely on specialized carrier-mediated transport systems, receptor-mediated transcytosis, and active efflux pumps embedded within the endothelial membrane 167.
Pericytes and Astrocytic Endfeet
Surrounding the endothelial layer are pericytes, which are specialized mural cells possessing contractility similar to smooth muscle cells 3. Pericytes act as essential gatekeepers of the microvasculature. They communicate directly with endothelial cells via physical peg-and-socket contacts and paracrine signaling pathways to regulate capillary blood flow, vessel maturation, and the expression of barrier-specific tight junction proteins 238. Furthermore, pericytes are primarily responsible for synthesizing critical extracellular matrix components, including type IV collagen and laminin, which form the foundational basement membrane 3.
Ensheathing the microvessels are the endfeet of astrocytes, the most abundant glial cells in the central nervous system. Astrocytic endfeet provide an additional physical boundary along the perivascular space and secrete specific signaling factors - such as sonic hedgehog, retinoic acid, angiopoietin-1, and glial cell line-derived neurotrophic factor - that promote the tightening of endothelial junctions 39. Astrocytes are also uniquely responsible for maintaining water, ion, and neurotransmitter homeostasis around the blood vessels, largely through the localized expression of aquaporin-4 water channels and glutamate transporters 10. Microglia, the resident immune cells of the central nervous system, constantly survey the neurovascular unit, ready to respond to injury or pathological breach by initiating localized inflammatory cascades or phagocytosing debris 311.
Molecular Composition of the Barrier Interface
The physical restriction of paracellular transport is achieved by highly specialized intercellular structures, namely tight junctions and adherens junctions. The spatial organization and functional capacity of these junctions are dynamically regulated by calcium-dependent signaling, phosphorylation events, and G-protein-mediated pathways, which govern their assembly, disassembly, and response to physiological stimuli 12.
Tight Junctions and Scaffolding Proteins
Tight junctions localize between adjacent endothelial cells, serving as the core elements actively involved in the establishment of the paracellular seal. These structures are formed by interacting transmembrane proteins, primarily the claudin family (with claudin-5 being the most prominent in the brain), occludin, and junctional adhesion molecules 5912. These transmembrane proteins interlock across the intercellular cleft, acting as an impenetrable gate. Inside the endothelial cell cytoplasm, the tight junction proteins are firmly anchored to the actin cytoskeleton by membrane-associated guanylate kinase scaffolding proteins, most notably zonula occludens-1 (ZO-1), ZO-2, and ZO-3 1012.
Additionally, proteins possessing the MARVEL domain - such as tricellulin and MARVELD3 - play critical roles in sealing the complex points where three adjacent endothelial cells meet 512. The loss or downregulation of these specific tight junction components immediately correlates with increased barrier permeability and subsequent neurovascular pathology 910.
The Dual-Layered Basement Membrane
The extracellular matrix surrounding the vascular endothelium, known as the basement membrane, is composed of two distinct biochemical layers. The inner endothelial basement membrane is secreted cooperatively by pericytes and endothelial cells. It consists of a crosslinked network of type IV collagen, nidogen, heparan sulfate proteoglycans (such as agrin and perlecan), and specific laminin isoforms (α4 and α5) 3589.
Conversely, the outer parenchymal basement membrane is secreted entirely by astrocytes and is compositionally distinct, containing α1 and α2 laminins 5. This dual-layered matrix provides critical structural support, anchors the cellular components via integrin receptors, and serves as an additional physical sieve against cellular infiltration. Type IV collagen specifically assists in maintaining the integrity of the barrier matrix, and its degradation by specific enzymes is a hallmark of barrier failure 39.
Pathogenic Mechanisms of Barrier Disruption
Disruption of the blood-brain barrier is not merely a downstream consequence of neurological disease; it is frequently a primary driver of pathology. When the barrier breaks down, the central nervous system is abnormally exposed to circulating neurotoxins, red blood cells, leukocytes, and blood-derived proteins such as fibrinogen, thrombin, and albumin 1313.
Enzymatic Degradation and Inflammatory Cascades
A central mechanism of barrier degradation involves the dysregulation of matrix metalloproteinases (MMPs), particularly MMP-9 and MMP-3. During states of neuroinflammation, traumatic injury, or ischemia, reactive oxygen species and pro-inflammatory cytokines - such as tumor necrosis factor-alpha and interleukin-1 beta - stimulate pericytes and resident microglia to release these proteolytic enzymes 11415. MMP-9 specifically cleaves the structural proteins of the basement membrane, including collagen IV, and degrades the tight junction proteins linking the endothelial cells 1415.
This degradation permits the influx of water molecules and blood components into the brain extracellular space, resulting in vasogenic brain edema 1617. Furthermore, compromised barriers allow peripheral macrophages and leukocytes to enter the brain parenchyma. This transmigration is facilitated by the formation of neutrophil extracellular traps, which interact with microglia to trigger inflammasome activation and cytokine release, creating a self-reinforcing cycle of chronic neuroinflammation 1617.
Transporter Failure and Hypoperfusion
Barrier breakdown often progresses through multiple interconnected stages rather than a single catastrophic failure. Early molecular dysfunction frequently involves the downregulation of critical nutrient transporters, such as the glucose transporter GLUT1, leading to diminished glucose transport and cellular energy crises 1213. Concurrently, the loss of amyloid-beta efflux pumps, specifically the low-density lipoprotein receptor-related protein 1 (LRP1), prevents the clearance of metabolic waste, promoting the accumulation of toxic proteins within the brain parenchyma 113.
Subsequently, as pericytes degenerate due to oxidative stress or toxic protein accumulation, the microvasculature loses its capillary tone. This leads to localized cerebral hypoperfusion, creating hypoxic conditions that further exacerbate endothelial damage 218. Finally, the physical unzipping of the tight junctions permits widespread paracellular leakage, culminating in the severe neurovascular uncoupling observed in advanced neurodegenerative states 21419.
Disease-Specific Phenotypes of Barrier Breakdown
Barrier dysfunction manifests differently depending on the specific disease etiology. Across ischemic, neurodegenerative, and traumatic pathologies, the timeline, anatomical location, and severity of barrier breakdown vary significantly, presenting unique clinical challenges and diagnostic markers.
| Neurological Condition | Primary Mechanism of Barrier Breakdown | Key Biomarkers & Pathological Features |
|---|---|---|
| Alzheimer's Disease | Pericyte degeneration, reduced LRP1/GLUT1 transport, Aβ vascular deposition (cerebral amyloid angiopathy). | Elevated CSF sPDGFRβ, extravascular fibrinogen/hemosiderin, early hippocampal gadolinium leakage (DCE-MRI). 121318 |
| Acute Ischemic Stroke | Hypoxia-induced MMP-9 activation degrading tight junctions and collagen IV. Leads to reperfusion injury. | Elevated blood MMP-9, high risk of hemorrhagic transformation, severe vasogenic edema, loss of ZO-1. 141720 |
| Multiple Sclerosis | Neuroinflammatory disruption facilitating T-cell, B-cell, and peripheral macrophage infiltration. | Elevated CSF MMP-9, perivascular cuffing, gadolinium leakage heavily localized to active white matter lesions. 1221 |
| Traumatic Brain Injury | Mechanical shearing of microvessels, secondary MMP-9 degradation, and severe astrocytic injury. | Elevated blood GFAP, UCH-L1, and S100B in acute phases (0-24 hours), potential progression to CTE. 1521 |
Alzheimer's Disease and Vascular Dementia
In Alzheimer's disease and associated vascular dementias, barrier breakdown is frequently characterized as an early, insidious process. Neuroimaging and post-mortem tissue analyses demonstrate that barrier leakage typically begins in the hippocampus, a brain region critical for learning and memory, and can precede the onset of hippocampal atrophy and measurable cognitive impairment 12223. This breakdown is strongly associated with pericyte degeneration and the deposition of amyloid-beta along the walls of blood vessels, a condition known as cerebral amyloid angiopathy 11824.
Individuals harboring the apolipoprotein E4 (APOE4) allele exhibit significantly thinner capillary basement membranes and increased plasma protein leakage into the cortex, independent of amyloid-beta accumulation 132526. The resultant influx of neurotoxic blood-derived debris initiates multiple pathways of neuronal injury and synaptic dysfunction 125.
Acute Ischemic and Hemorrhagic Stroke
In acute ischemic stroke, blood-brain barrier disruption follows a specific temporal and mechanistic pattern that limits the therapeutic time window for clinical intervention. While the barrier remains largely intact for the first two hours following an ischemic event, severe disruption is typically observed between 6 and 48 hours post-onset, corresponding with peak MMP-9 activity 1417.
This disruption is the primary risk factor for hemorrhagic transformation - a severe, often fatal complication where blood extravagates into the brain tissue following the administration of tissue plasminogen activator (tPA) or mechanical thrombectomy 172027. Restoring blood flow is necessary to salvage the ischemic penumbra, but reperfusing vulnerable microvessels that have lost their tight junction integrity frequently results in catastrophic vascular rupture 1427. In contrast, primary hemorrhagic stroke directly obliterates the local microvasculature, triggering immediate, widespread barrier permeability and massive neuroinflammatory responses driven by the sudden presence of extravascular erythrocytes 1617.
Traumatic Brain Injury and Multiple Sclerosis
Following a traumatic brain injury (TBI), the immediate mechanical shearing of microvessels causes acute structural damage to the barrier. In the subacute and chronic phases, secondary injury mechanisms driven by MMPs and neuroinflammation sustain barrier permeability, contributing to post-traumatic cerebral edema 15. Repeated mild TBIs can lead to chronic traumatic encephalopathy (CTE), characterized by persistent perivascular space enlargement and the accumulation of hemosiderin-laden macrophages around deep penetrating vessels 1.
In multiple sclerosis, barrier disruption is an essential prerequisite for disease progression. The pathology centers on the targeted degradation of endothelial tight junctions, such as ZO-1, which permits autoimmune lymphocytes and macrophages to cross the barrier and attack the myelin sheaths of neurons 12. Active demyelinating plaques in the white matter consistently exhibit severe local barrier leakage and persistent fibrinogen extravasation 12.
Fluid and Neuroimaging Biomarkers of Barrier Integrity
The ability to measure blood-brain barrier breakdown in vivo has dramatically improved with the advent of advanced neuroimaging protocols and highly sensitive fluid biomarker assays. Historically, assessing barrier integrity required invasive procedures or was strictly limited to post-mortem histological analysis 118.
Advanced Neuroimaging Techniques
Dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) utilizing gadolinium-based contrast agents is currently the gold standard for quantifying barrier permeability in the living human brain 12228. By measuring the rate of contrast extravasation from the intravascular space into the brain parenchyma, clinicians can calculate the Ktrans permeability constant, mapping subtle, regional barrier leaks 2228.
In conditions like mild cognitive impairment and early Alzheimer's disease, DCE-MRI has consistently revealed that barrier breakdown begins in the hippocampus prior to the onset of measurable brain atrophy 12223. Arterial spin labeling (ASL) MRI techniques are also emerging as valuable tools, allowing for the evaluation of barrier integrity based on different diffusion or transverse relaxations without the need for exogenous contrast agents 1729. Furthermore, positron emission tomography (PET) using specific radiotracers can measure the functional impairment of the barrier, identifying diminished glucose transport (via FDG-PET) or the loss of efflux pump activity 122.
Fluid Biomarkers in Blood and Cerebrospinal Fluid
The transition toward blood- and cerebrospinal fluid (CSF)-based biomarkers provides a rapid, less invasive methodology for risk stratification, diagnostics, and longitudinal monitoring. The classic metric for generalized barrier leakiness is the albumin quotient (Qalb), which measures the ratio of albumin in the CSF relative to the blood serum 12229. Because albumin is exclusively synthesized in the liver, its presence in the CSF directly reflects the severity of barrier permeability, a finding consistently elevated in multiple sclerosis and severe dementia 122.
More granular cellular biomarkers have recently been validated for clinical use. Soluble platelet-derived growth factor receptor beta (sPDGFRβ) has emerged as a highly specific indicator of pericyte injury. Elevated levels of sPDGFRβ in the CSF strongly correlate with regional barrier breakdown in the hippocampus, independent of amyloid or tau biomarker status, making it a critical early indicator of neurovascular decline 122326.
For acute trauma and stroke, glial fibrillary acidic protein (GFAP) and ubiquitin C-terminal hydrolase L1 (UCH-L1) are heavily utilized. GFAP is an intermediate filament protein localized within astrocytes. When astrocytic endfeet are damaged or the barrier undergoes mechanical shear stress, GFAP is rapidly released into the systemic circulation, serving as an exceptional diagnostic marker for identifying acute intracranial injury and determining the necessity of CT imaging 101521. UCH-L1 is a neuronal enzyme linked to axonal damage and synaptic dysfunction. When measured in the blood alongside GFAP, it provides strong prognostic value in predicting mortality and global functional recovery at six months post-injury 101521. Additional emerging markers for neuroinflammation and neurovascular unit disruption include matrix metalloproteinase-9 (MMP-9), neurofilament light chain (NfL), S100B, and YKL-40, which are increasingly incorporated into multi-marker diagnostic panels 10212830.
Evolving Scientific Consensus on Neurovascular Pathology
The specific role and timeline of blood-brain barrier failure in Alzheimer's disease pathophysiology has been a subject of intense recent scrutiny. The landscape of neurovascular research was heavily disrupted in late 2023 and 2024 by highly publicized scientific investigations, allegations of extensive data manipulation, and high-profile retractions.
The Zlokovic Investigation and Clinical Trial Suspensions
For over a decade, the dominant theory emphasizing the primacy of pericyte degeneration and early vascular leakage in Alzheimer's disease was spearheaded by prominent researcher Berislav Zlokovic of the University of Southern California (USC) 3132. His laboratory generated foundational hypotheses suggesting that barrier dysfunction precedes and actively drives the accumulation of amyloid-beta, rather than occurring simply as a downstream effect 31. This body of work directly underpinned the development of 3K3A-APC, a modified version of human activated protein C. Preclinical and phase II data suggested that 3K3A-APC could protect barrier integrity, reduce neuroinflammation, and significantly lower the risk of hemorrhagic transformation following stroke therapy 3334.
However, in late 2023, an independent group of whistleblowers submitted a 113-page dossier to the National Institutes of Health (NIH) alleging widespread data and image manipulation in 35 papers authored by Zlokovic, including the foundational preclinical data supporting 3K3A-APC 353637. Crucially, the whistleblowers alleged that the phase II clinical trial data for 3K3A-APC may have been manipulated to obscure adverse outcomes, noting a potentially higher rate of early mortality and worse disability among patients receiving the drug compared to the placebo group 3738.
In response, the NIH immediately paused the planned $30 million phase III clinical trial and demanded the return of preliminary funding 383940. The corporate sponsor, ZZ Biotech, formally withdrew the trial from government registries in late 2024, stating that the future of the drug's development relies on the outcome of ongoing investigations 36. Subsequently, USC placed Zlokovic on indefinite leave, and several major scientific journals began issuing retractions, corrections, and expressions of concern regarding his published papers 383940. This upheaval was compounded by parallel findings of data fabrication that led to the ouster of Eliezer Masliah, the head of the neuroscience division at the National Institute on Aging, further rocking the neurodegeneration research community 41.
Recalibrating the Alzheimer's Disease Models
These controversies have forced the broader scientific community to critically re-evaluate the vascular hypothesis in Alzheimer's disease. While the correlation between generalized vascular dysfunction and dementia remains an accepted clinical reality, recent independent studies challenge the previously accepted scale and physical nature of barrier degradation.
A pivotal 2025 study from the Texas Tech University Health Sciences Center evaluated the Tg2576 Alzheimer's mouse model using advanced pharmacokinetic analysis and stable isotope-labeled sucrose tracers. The researchers found no widespread paracellular leakage across the barrier, demonstrating that sucrose levels in the brain remained extremely low 2442. Furthermore, immunohistochemical analysis showed that tight junction proteins (claudin-5, occludin, ZO-1) remained structurally sound even in the immediate vicinity of heavy vascular amyloid plaques, fundamentally challenging the assumption that Alzheimer's disease causes gross, widespread physical leakiness of the microvasculature 2442.
Simultaneously, a 2025 study published in Neuron by the Gladstone Institutes shifted the focus away from structural ruptures toward complex genetic regulation. The study demonstrated that the vast majority of genetic risk factors associated with Alzheimer's disease and stroke do not act within neurons, but rather exert their effects primarily within the vascular and immune "border cells" that form the barrier 43. Consequently, the scientific consensus is rapidly evolving: rather than a gross physical rupturing of the barrier in Alzheimer's disease, the primary pathology may be defined by highly localized microvascular failures, altered transporter functions, and aberrant genetic signaling within the neurovascular unit's constituent cells 222443.
Therapeutic Bypassing and Restoration Mechanisms
Because the healthy blood-brain barrier effectively excludes nearly 100% of large-molecule neurotherapeutics and over 98% of small-molecule drugs, achieving effective pharmacological delivery remains a monumental hurdle in the treatment of central nervous system disorders 444546. Current clinical research aims to either exploit the barrier's endogenous transport systems or safely, transiently disrupt the tight junctions to allow therapeutic compounds to pass.
Receptor-Mediated and Transporter-Mediated Transcytosis
Receptor-mediated transcytosis (RMT) utilizes the barrier's own carrier systems to shuttle large biotherapeutics into the brain. Endothelial cells heavily express specific receptors - such as the transferrin receptor (TfR), the insulin receptor, and the low-density lipoprotein receptor - that naturally bind to circulating physiological ligands, package them into intracellular vesicles, and transport them across the cytoplasm for release into the brain parenchyma 6746.
Pharmaceutical researchers engineer "molecular Trojan horses" by fusing a therapeutic payload (such as an enzyme, gene plasmid, or antisense oligonucleotide) to a monoclonal antibody specifically designed to bind to these endothelial receptors 6747. A landmark breakthrough occurred recently with the regulatory approval in Japan of pabinafusp alfa, a brain-penetrating fusion protein targeting the transferrin receptor to deliver iduronate-2-sulfatase 64648. This therapy successfully treats Hunter syndrome (mucopolysaccharidosis type II), addressing severe neurological symptoms that conventional, non-penetrating enzyme replacement therapies could not reach 646. A second generation of these biotherapeutics is rapidly advancing, with clinical trials currently evaluating valanafusp alpha for Hurler's syndrome and trontinemab for Alzheimer's disease 648. Transporter-mediated transcytosis (TMT) is a parallel strategy that exploits endogenous solute carriers, such as the glucose transporter GLUT1, to pull targeted nanocarriers across the endothelium 47.
Focused Ultrasound and Nanoparticle Delivery
For targeted, region-specific drug delivery, transcranial focused ultrasound (FUS) combined with intravenously administered gas-filled microbubbles offers a highly controlled, non-invasive method of transient barrier opening 454950.
When targeted low-frequency acoustic waves are applied to a predetermined coordinate within the brain, the circulating microbubbles oscillate stably within the focal region. This oscillation generates localized acoustic radiation forces and mechanical stress on the vessel wall, which safely and reversibly disassembles the tight junctions for several hours 495051.
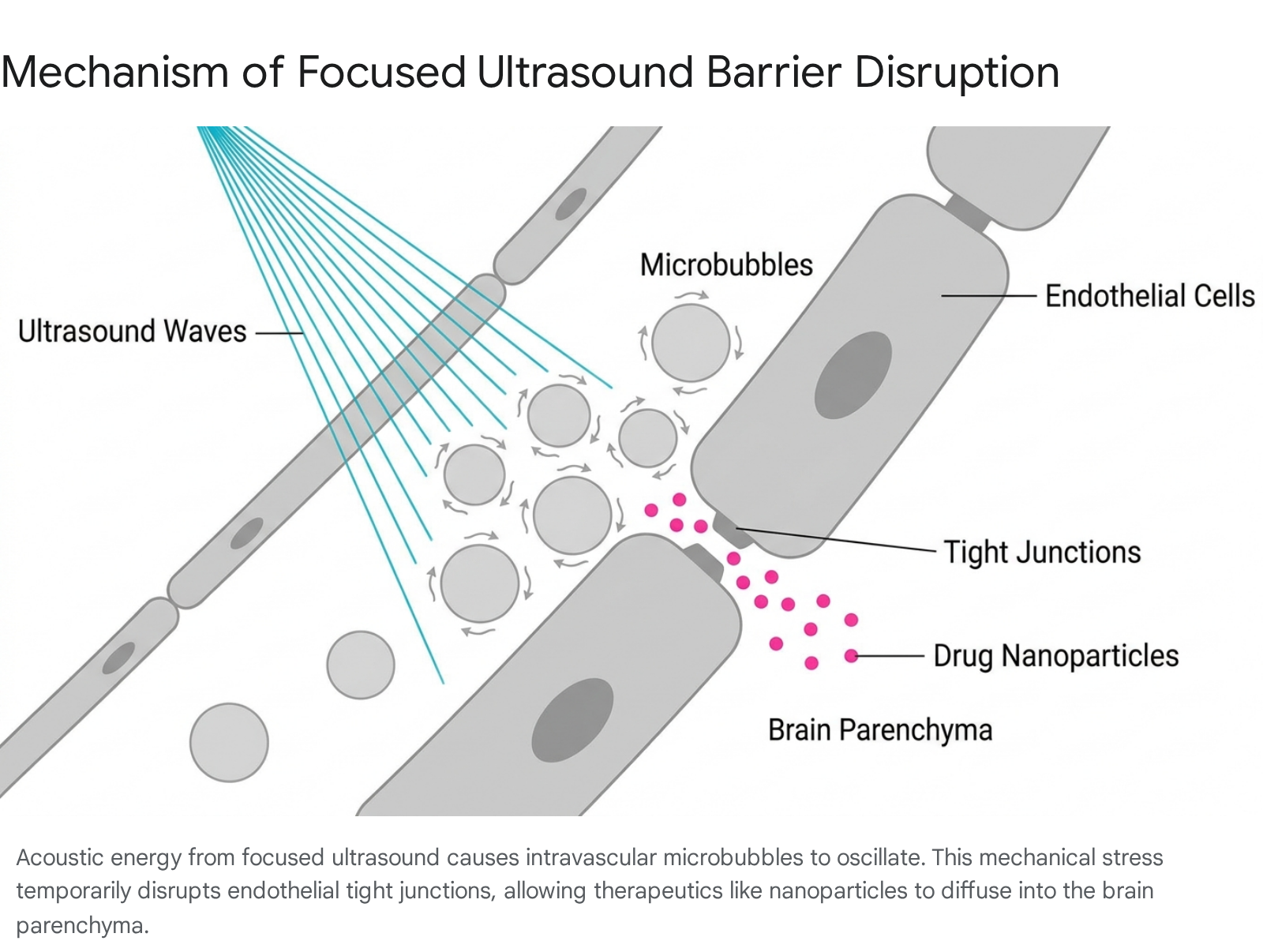
This technique has been successfully utilized in human clinical trials to enhance the systemic delivery of albumin-bound paclitaxel to recurrent glioblastomas 454950. Preclinical studies show immense promise for pairing FUS with highly engineered lipid nanoparticles (LNPs) and polymeric nanoparticles. In murine models, FUS-mediated opening has enabled the robust, non-invasive delivery of LNPs loaded with mRNA and siRNA directly into brain tumors and specific brain parenchymal cells, achieving high levels of targeted gene transfer and somatic genome editing without causing permanent vascular damage or provoking systemic toxicity 495051.
Global Research Consortiums
The complexities of navigating the blood-brain barrier have necessitated massive, international collaborative efforts. The IM2PACT consortium, backed by the Innovative Medicines Initiative (IMI) and coordinated by the University of Oxford and Sanofi, brings together 27 global partners to develop highly predictable in vitro stem cell models (iPSC lines) of the human barrier and discover novel brain delivery systems 5253. Similarly, in the Asia-Pacific region, neurointerventional advancements are heavily driven by collaborative platforms such as the East Asian Conference on Neurointervention (EACoN) and the Asian Network for Research Resource Centers (ANRRC), which foster the exchange of cutting-edge research, minimally invasive surgical techniques, and biological resource sharing among institutions in South Korea, China, and Japan 54555657.
Clinical Reality Versus Wellness Industry Interventions
As rigorous scientific research into blood-brain barrier permeability has accelerated, the underlying concepts have been aggressively co-opted by the global alternative health and functional medicine industries. Patterned closely after the scientifically dubious "leaky gut syndrome," the wellness industry frequently markets "leaky brain syndrome" as a catch-all diagnostic root cause for brain fog, chronic fatigue, mild depression, and attention deficits 585960.
Evaluating "Leaky Brain" Supplements
The global wellness industry generates billions in revenue from direct-to-consumer supplements claiming to "heal" barrier permeability, enhance memory, and reverse cognitive decline 616263. These over-the-counter formulations routinely feature ingredients such as omega-3 fatty acids, ginkgo biloba, B-complex vitamins, vitamin D, vitamin E, and various botanical extracts 636465.
However, regulatory agencies such as the FDA do not govern these dietary supplements for clinical efficacy, and established medical authorities, including the National Institutes of Health and the Mayo Clinic, find no compelling clinical evidence to support their use for curing barrier permeability or preventing dementia in healthy individuals 636667. Exhaustive, long-term clinical trials have repeatedly failed to demonstrate efficacy. For example, the Ginkgo Evaluation of Memory (GEM) study, which tracked over 3,000 older adults over six years, conclusively demonstrated that ginkgo biloba offers no protective effect against cognitive decline or the onset of dementia compared to a placebo 6468. Similarly, while severe clinical deficiencies in vitamins B6, B9, or B12 can elevate homocysteine and mimic cognitive dysfunction, supplementing these vitamins in non-deficient populations yields no statistically significant preventative benefit against Alzheimer's disease or vascular dementia 6368.
Preclinical Promise versus Human Bioavailability
This lack of observable clinical efficacy in humans stands in stark contrast to specific in vitro and animal models, where certain isolated nutraceuticals demonstrate legitimate, potent biochemical activity. Dietary compounds such as resveratrol (derived from grape skins), curcumin (from turmeric), berberine, fucoidan, and specific urolithins have been shown in highly controlled preclinical models to exert significant anti-inflammatory and antioxidant effects on brain endothelial cells 69707172.
In rodent models of ischemia and major depressive disorder, these compounds can reduce astrocyte swelling and upregulate crucial cellular survival pathways (including WNT, PI3K-AKT, and NRF2) while suppressing inflammatory signaling cascades (such as MAPK, ERK, and NF-κB) 697172. This biochemical modulation effectively tightens intercellular junctions, reduces MMP-9 activity, and limits vasogenic edema in the animal models 6972.
However, translating these preclinical mechanisms into human therapies is severely limited by pharmacokinetics. These results rely on precise, isolated, and often intravenous dosing conditions that are practically unachievable through standard oral commercial supplements due to the compounds' inherently low systemic bioavailability, rapid metabolism, and poor natural penetration across the intact human barrier 7172. Consequently, the overarching medical consensus remains that standard lifestyle interventions - specifically regular aerobic exercise, strict management of cardiovascular risk factors (like hypertension), intellectual stimulation, and adherence to a nutrient-dense Mediterranean-style diet - are vastly superior, evidence-based strategies for maintaining neurovascular unit health and delaying cognitive decline compared to any currently available over-the-counter supplement 6467.