Pathophysiology of age-related chronic low-grade inflammation
Introduction
The conceptualization of aging has undergone a profound transformation, moving from a stochastic model of inevitable entropy to a heavily regulated, modifiable biological trajectory. Central to this paradigm shift is the concept of "inflammaging," a term originally coined by Claudio Franceschi in 2000 to describe the chronic, sterile, low-grade systemic inflammation that acts as a fundamental driver of organismal senescence and age-related morbidities 123. While acute inflammation is a highly coordinated, self-limiting biological response necessary for pathogen neutralization and tissue repair, inflammaging represents a fundamentally dysregulated, smoldering immune activation.
It is vital to explicitly distinguish chronic sterile inflammation from necessary acute responses. Acute inflammation relies on transient signaling - where pathogen-associated molecular patterns (PAMPs) or localized tissue damage initiate a rapid innate immune response that is subsequently resolved by counter-regulatory cytokines (such as IL-10 and TGF-$\beta$) and specialized pro-resolving lipid mediators 35. Once the exogenous stressor is neutralized, tissue homeostasis is fully restored. In stark contrast, inflammaging is a sterile phenomenon, devoid of overt infection, and entirely non-resolving. It is driven by the continuous accumulation of endogenous damage-associated molecular patterns (DAMPs), cellular debris, mitochondrial dysfunction, and the accumulation of senescent cells across multiple organ systems. This constant low-level activation of the innate immune system simultaneously impairs adaptive immune competence - a process termed immunosenescence - blunting vaccine efficacy and creating a highly pro-inflammatory microenvironment that serves as the common soil for age-related chronic diseases 147.
The scientific consensus increasingly recognizes that inflammaging is not an isolated hallmark of aging but a biological nexus. It is intimately interconnected with genomic instability, telomere attrition, epigenetic drift, loss of proteostasis, and mitochondrial dysfunction 47. Recent breakthroughs in precision geroscience - particularly human demographic and molecular data emerging between 2023 and 2026 - have expanded the understanding of inflammaging across diverse populations, revealing that it may not be a universal biological inevitability, but rather an evolutionary mismatch severely exacerbated by industrialized environments 56. Concurrently, the rigorous exploration of sleep microarchitecture, exercise-induced myokines, the gut virome and microbiome, and advanced senotherapeutic pharmacology has opened unprecedented avenues for clinical intervention.
Biological Drivers and the Senescence-Associated Secretory Phenotype (SASP)
The physiological transition from a youthful, homeostatic state to an aged, inflamed phenotype is governed by specific cellular mechanisms operating at the epigenetic and transcriptional levels. Cellular senescence - a state of stable, terminal cell-cycle arrest triggered by intrinsic and extrinsic stressors such as DNA damage, oncogene activation, and oxidative stress - acts as the primary biological engine of inflammaging. While senescent cells permanently cease to divide, they remain highly metabolically active, secreting a complex, deleterious cocktail of pro-inflammatory cytokines, chemokines, growth factors, and proteases collectively known as the Senescence-Associated Secretory Phenotype (SASP) 789.
Transcriptional Reprogramming and SASP Propagation
The physical clustering of SASP genes and broader changes in chromatin conformation underscore a profound epigenetic reprogramming in senescent cells 8. The relocation of specific histone variants, such as the macroH2A1 histone variant, away from SASP genes following endoplasmic reticulum (ER) stress enables the massive transcriptional upregulation of inflammatory mediators 8. Crucially, key SASP components, most notably Interleukin-6 (IL-6), Interleukin-8 (IL-8), GRO$\alpha$, and Insulin-like Growth Factor-Binding Protein 7 (IGFBP-7), operate in powerful autocrine positive feedback loops 78. These self-sustaining feedback mechanisms reinforce the senescent state, ensuring its persistence even after the initial cellular stressor has dissipated.
Furthermore, the SASP exhibits potent paracrine effects, initiating a phenomenon known as secondary or bystander senescence 810. For instance, senescent human oral epithelial cells secrete factors that induce senescence-associated $\beta$-galactosidase (SA-$\beta$-gal) activity and SASP expression in neighboring healthy keratinocytes 7. The temporal regulation of the SASP is distinctly biphasic. The early phase is characterized by the hyper-secretion of interleukins (IL-6, IL-8, IL-1$\alpha$, IL-1$\beta$) driven by NF-$\kappa$B and p38MAPK signaling pathways 7810. This is followed by a protracted late phase characterized by the profound expression of extracellular matrix (ECM) modulators and profibrotic factors 7810.
Table 1: Biological Drivers and SASP Components in Inflammaging
| Component Category | Key Molecules | Pathophysiological Impact | Source Relevance |
|---|---|---|---|
| Pro-inflammatory Cytokines | IL-6, IL-1$\alpha$, IL-1$\beta$, TNF-$\alpha$ | Establish autocrine senescence circuits; induce paracrine bystander senescence; drive systemic metaflammation and inhibit adaptive immune responses. | 78910 |
| Chemokines | IL-8 (CXCL8), CCL2, CCL20 | Recruit innate immune cells (macrophages, neutrophils) to tissues; promote aberrant tissue remodeling and microenvironment immunosuppression. | 78910 |
| Proteolytic Enzymes | MMP-1, MMP-3, MMP-12 | Degrade collagen and ECM structures; compromise tissue barriers; facilitate epithelial-mesenchymal transition (EMT) and tumor invasion. | 78 |
| Growth Factors & Modulators | TGF-$\beta$, GDF15, PAI-1, CST3, IGFBP2 | TGF-$\beta$ and PAI-1 act as primary drivers of tissue fibrosis; GDF15, CST3, and IGFBP2 form a highly robust biomarker cluster linked to hematological decline and renal dysfunction. | 27914 |
Matrix Metalloproteinases (MMPs) constitute a highly destructive element of the late-phase SASP. By degrading collagen directly or activating downstream fibrinolytic protease cascades, MMPs destroy connective tissues, leading to clinical pathologies ranging from periodontal disease to aggressive vascular degradation 7. Additionally, Transforming Growth Factor-beta (TGF-$\beta$) acts in concert with other profibrotic cytokines (such as endothelin and CTGF) and tissue inhibitors of metalloproteinases (TIMP-1 and -2), driving progressive tissue fibrosis 79. This fibrotic signaling replaces functional parenchyma with non-functional scar tissue, a fundamental hallmark of aging in the heart, lungs, and liver. Concurrently, elevated levels of Plasminogen Activator Inhibitor-1 (PAI-1/SERPINE1) progressively accumulate in systemic circulation, further driving cellular senescence, tissue remodeling, and cardiovascular pathogenesis 78.
The Pathophysiological Axis: Inflammaging and Age-Related Disease
Because circulating levels of SASP factors operate systemically, inflammaging is pleiotropic, affecting diverse organ systems simultaneously and directly fueling the pathogenesis of the most prevalent diseases of aging.
The Tumor Microenvironment and Oncogenesis
Inflammaging creates a highly permissive microenvironment for oncogenesis. The classical view of senescence as a purely tumor-suppressive mechanism - by halting the proliferation of cells with damaged DNA - has been thoroughly revised. While initially protective, the chronic persistence of senescent cells and their SASP secretome strongly promotes tumorigenesis in surrounding tissues 910.
The IL-6/JAK/STAT3 signaling pathway serves as a critical axis in this malignant transformation. Elevated IL-6 levels in the tumor microenvironment stimulate tumor cell proliferation, survival, and metastasis 910. Moreover, IL-6 induces profound immunosuppression by activating bone marrow-derived myeloid suppressor cells (MDSCs) and causing the depletion and exhaustion of CD8+ cytotoxic T cells, effectively blinding the host immune system to malignant antigens 910. SASP factors, particularly IL-6 and IL-8, also trigger the epithelial-mesenchymal transition (EMT), granting cancer cells enhanced invasive capabilities, facilitating basement membrane invasion, and contributing to therapeutic resistance and recurrence following targeted oncotherapy 910.
Sarcopenia, Frailty, and Myosteatosis
In the musculoskeletal system, inflammaging directly accelerates sarcopenia - the progressive, catabolic decline in skeletal muscle mass, strength, and overall functional reserve. Sarcopenia is driven by elevated levels of pro-inflammatory cytokines, which tilt the metabolic balance away from protein synthesis and toward muscle degradation 1112. A heavily under-recognized component of this physical decline is myosteatosis, characterized by the pathological infiltration of adipose tissue into skeletal muscle architectures 12. Ectopic lipid deposition in muscle not only compromises mechanical force generation but also serves as a localized source of inflammatory adipokines, exacerbating insulin resistance and local tissue inflammation. Despite its high prevalence across metabolically vulnerable populations and cancer survivors, a lack of standardized imaging consensus cutoffs continues to hinder the cross-study evaluation of myosteatosis as an early biomarker of frailty 12.
Neurodegeneration, Autoimmunity, and Immune Dysfunction
In the central nervous system, systemic inflammaging crosses the blood-brain barrier, activating local microglia and astrocytes. This neuroinflammation disrupts synaptic homeostasis, impairs the brain's glymphatic clearance system, and promotes the persistent aggregation of neurotoxic proteins, such as amyloid-$\beta$, tau, and $\alpha$-synuclein 417. Sustained activation of the NLRP3 inflammasome and NF-$\kappa$B signaling within glial cells accelerates neuronal apoptosis and is independently linked to the progression of Alzheimer's disease and other neurodegenerative conditions 417. Similarly, inflammaging severely impacts mucosal immunity, altering the pathogenesis of conditions like Inflammatory Bowel Disease (IBD). In late-onset IBD, immunosenescence and elevated systemic inflammatory tone reshape the host response to intestinal microbiota, resulting in diminished T-cell diversity and prolonged disease activity 13. Systemic immune responses also alter, evidenced by the "Treat-to-Target" recommendations in Rheumatoid Arthritis (RA), where frailty assessments dictate that while non-frail older adults require aggressive management, frail patients require highly individualized therapeutic care due to diminished organ resilience 19.
The Centenarian Paradox and Genomic Admixture
A central question in precision geroscience is why some individuals - centenarians - achieve extreme longevity while escaping or significantly delaying the onset of inflammaging and its associated diseases. The study of exceptional longevity in geographically diverse "Blue Zones" provides vital clues into the genetic, epigenetic, and environmental modulators of the aging immune system.
Genetic Architecture in Okinawa and Nicoya
In Okinawa, Japan, where the centenarian prevalence reaches extraordinarily high levels (40 - 50 per 100,000 persons), genomic research has identified a distinct clustering of longevity-associated gene variants characteristic of a founder population 142115. The FOXO3 gene, a master transcriptional regulator involved in apoptosis, oxidative stress resistance, and insulin signaling, shows significant allelic variations (specifically SNP rs2802292) that are highly conserved among Okinawan centenarians 2116. Furthermore, specific Human Leukocyte Antigen (HLA) alleles in this population provide natural protection against autoimmune and inflammatory dysregulation, effectively dampening the lifetime burden of systemic inflammation. Additionally, unique mitochondrial gene variants, such as Mt5178A, contribute to optimized metabolic function 21.
Similarly, the Nicoya Peninsula of Costa Rica presents a profound survival advantage, particularly for males, who demonstrate a death rate ratio (DRR) of 0.80 compared to the rest of the country, and a specifically lowered cardiovascular mortality risk (DRR = 0.65) 1725. Biological markers of aging in Nicoyans reflect exceptionally high physiological resilience to inflammaging. Nicoyans possess unusually long leukocyte telomere lengths (LTL) - averaging 81 base pairs longer than age-matched cohorts in other regions - serving as a molecular marker of reduced cumulative cellular stress 1819. They also exhibit favorable levels of dehydroepiandrosterone sulfate (DHEA-S) and lower levels of CD8+ T cells, indicating preserved immune regulation 1718. Intriguingly, genomic analyses have linked this extraordinary longevity to a higher proportion of Amerindian genetic ancestry, with each 10% increase in Amerindian admixture corresponding to a 2.32-fold greater likelihood of reaching centenarian status 20. Furthermore, youthful DNA methylation patterns identified in Nicoyan elders have also been corroborated in centenarians from Sardinia and Ikaria, suggesting shared epigenetic resilience mechanisms across distinct geographic isolates 18.
The Paradigm Shift: Inflammaging as an Evolutionary Mismatch
Historically, inflammaging was viewed as a universal, inescapable biological law of human aging. However, a landmark 2025 study published in Nature Aging fundamentally disrupted this assumption by assessing immune profiles across four geographically and socioeconomically distinct populations 56. The study compared two highly industrialized cohorts (the Italian InCHIANTI study and the Singapore Longitudinal Aging Study) against two indigenous, non-industrialized forager-horticulturalist populations: the Tsimane of the Bolivian Amazon and the Orang Asli of Peninsular Malaysia 56.
The immunological findings were revelatory. In the industrialized populations, a clear "inflammaging axis" was identified, wherein baseline levels of cytokines (such as IL-6 and TNFR1) rose steadily and pathologically with chronological age, correlating strongly with chronic disease incidence 356. However, in the Tsimane and Orang Asli populations, this trajectory was virtually absent.
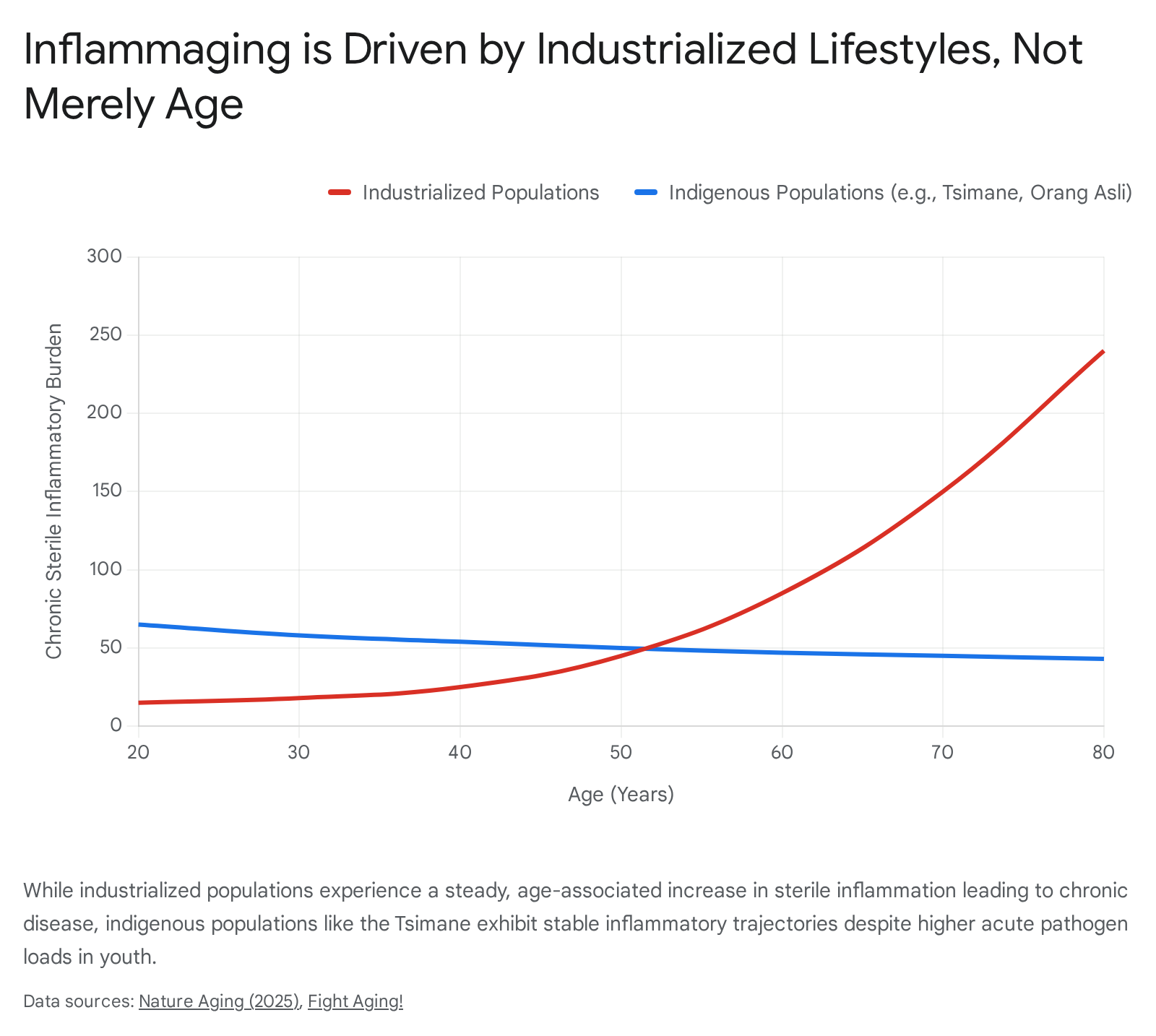
Due to high endemic pathogen loads and helminth infections, these indigenous groups exhibited high constitutive levels of systemic inflammation during youth 76. Crucially, however, these inflammatory markers did not undergo an age-dependent, chronic escalation, nor did they translate into age-related morbidities in later life 76. Chronic diseases like atherosclerosis, type 2 diabetes, and Alzheimer's disease remain remarkably rare in these populations 7.
This comprehensive data leads to a profound paradigm shift: inflammaging is not an obligatory hallmark of human biology, but rather a context-dependent evolutionary mismatch 529. It appears to be largely a byproduct of industrialized lifestyles - characterized by highly processed diets, caloric surplus, sedentary behavior, and the total absence of natural parasitic immunomodulation - interacting with a human physiology that evolved to manage acute, high-stress infectious challenges 2921.
Modulators of Inflammaging: Sleep Architecture and Exercise Immunology
Beyond genetic and geographic factors, specific behavioral and lifestyle interventions possess profound immunomodulatory capabilities, directly influencing the molecular pathways that govern systemic inflammaging.
Sleep Architecture: The Slow-Wave Sleep Imperative
The architecture of human sleep undergoes dramatic, deleterious deterioration with advancing age. Specifically, Slow-Wave Sleep (SWS), defined as the deep N3 sleep stage characterized by low-frequency delta waves, decreases by approximately 75% between the ages of 20 and 60 3122. SWS is not merely a period of rest; it is a highly active metabolic state critical for systemic homeostasis, neuroendocrine regulation, and immune resolution 2223.
During SWS, the systemic secretion of Growth Hormone (GH) peaks - a restorative process that declines by 80% alongside the loss of deep sleep in aging adults - while cortisol levels are heavily suppressed 31. The structural loss of SWS directly correlates with increased intracellular production and systemic circulation of pro-inflammatory cytokines, specifically IL-6 and TNF-$\alpha$ 2425. Experimental disruption of SWS via forced awakening in healthy adults induces a rapid upregulation of Toll-like receptor-4 (TLR-4) stimulated monocyte production of IL-6 25. This cellular inflammatory spike manifests clinically as heightened daytime fatigue, increased heat pain sensitivity (hyperalgesia), and profoundly impaired glucose homeostasis 242526. In vulnerable patient populations, such as children and adolescents with Major Depressive Disorder (MDD), reduced SWS and an increased proportion of lighter N2 sleep is significantly correlated with elevated high-sensitivity C-reactive protein (hsCRP) and severe insomnia 27.
Furthermore, SWS is required for the optimal functioning of the glymphatic system, the brain's macroscopic waste clearance pathway. The age-related loss of delta-wave power during NREM sleep directly hinders the clearance of metabolic byproducts, triggering localized neuroinflammation and the subsequent downstream activation of systemic inflammaging cascades 1723. The molecular response to this chronic physiological stress is further modulated by the Conserved Transcriptional Response to Adversity (CTRA), wherein prolonged stress down-regulates anti-viral gene expression and heavily up-regulates pro-inflammatory NF-$\kappa$B transcription factors 38. This phenomenon is closely tied to the prolonged induction of the transcription factor $\Delta$FosB in the nucleus accumbens, acting as both a marker of chronic neuronal activation and a mediator of long-term stress adaptation 39.
Exercise Immunology and the Myokine Paradox
Skeletal muscle is increasingly recognized not just as a mechanical organ required for locomotion, but as a primary endocrine organ. During rigorous contraction, muscle fibers synthesize and secrete bioactive peptides and cytokines known as myokines, which exert profound systemic anti-inflammatory effects and facilitate inter-organ crosstalk 2829. The biology of myokines presents a fascinating physiological paradox regarding IL-6. While macrophage-derived IL-6 is a key driver of the pro-inflammatory SASP, exercise-induced, muscle-derived IL-6 operates in a strictly anti-inflammatory context.
Following resistance or aerobic training, acute elevations in muscle-derived IL-6 stimulate systemic glucose uptake and lipolysis without activating pathological pro-inflammatory pathways. Crucially, muscle-derived IL-6 actively inhibits the production of TNF-$\alpha$ and stimulates the release of potent anti-inflammatory mediators, including Interleukin-1 receptor antagonist (IL-1ra) and Interleukin-10 (IL-10) 2830.
Other critical myokines upregulated during physical activity include Leukemia Inhibitory Factor (LIF), Interleukin-15 (IL-15), Fibroblast Growth Factor 2 (FGF2), FGF21, and Insulin-like Growth Factor-1 (IGF-1) 2830. Of particular note is Irisin, which is cleaved from the FNDC5 membrane protein. Irisin drives the "browning" of white adipose tissue to enhance thermogenesis and crosses the blood-brain barrier to directly induce the expression of Brain-Derived Neurotrophic Factor (BDNF) via Tropomyosin receptor kinase B (TrkB) signaling 3031. This irisin-BDNF axis enhances hippocampal neurogenesis, improves synaptic plasticity, and mitigates neuroinflammation, providing a robust molecular mechanism for the neuroprotective effects of exercise in mitigating Parkinson's and Alzheimer's disease progression 3031. By ensuring the maintenance of functional muscle mass and maximizing myokine output, structured resistance training serves as one of the most effective non-pharmacological interventions to actively suppress inflammaging 1129.
The Gut-Immune Axis and Metaflammation
The concept of "metaflammation" - metabolically triggered inflammation - expands the classical definition of inflammaging by directly linking modern dietary habits, obesity, and profound gut microbiota dysbiosis to accelerated biological aging 121. As humans age, the gut microbiome (GM) undergoes predictable, pathological structural shifts: a massive loss of overall species diversity, a severe depletion of beneficial immunomodulatory commensals (such as Bifidobacterium), and a massive expansion of pro-inflammatory Proteobacteria 1332.
This pervasive dysbiosis compromises intestinal barrier integrity. The degradation of the mucosal barrier allows lipopolysaccharides (LPS) and microbial metabolites to leak continuously into systemic circulation - a condition known clinically as metabolic endotoxemia 3245. Circulating LPS binds to TLR-4 receptors on circulating immune cells, triggering continuous NF-$\kappa$B activation and feeding the inflammaging cycle. Beyond bacteria, dysbiosis of the gut virome (reduced viral diversity and altered phage-bacteria interactions) and mycobiota further amplify this inflammatory signaling and impair metabolic homeostasis 45.
Emerging research from 2025 and 2026 highlights that therapies aggressively targeting GM restoration, such as Fecal Microbiota Transplantation (FMT), provide profound systemic benefits. Preclinical critical care models have demonstrated that FMT not only repairs intestinal barrier function by heavily upregulating tight junction proteins (ZO-1 and occludin) but also drastically reduces pulmonary pathological damage and systemic levels of TNF-$\alpha$, IL-6, and IL-1$\beta$ 45. Crucially, FMT has been shown to ameliorate systemic mitochondrial dysfunction by rebalancing mitochondrial dynamics - increasing the expression of fusion proteins (OPA1, Mfn1, Mfn2) while decreasing the expression of fission proteins (Drp1), thereby reducing oxidative stress 45. Furthermore, the deployment of Glucagon-Like Peptide-1 (GLP-1) receptor agonists, initially indicated solely for type 2 diabetes and metabolic syndrome, exerts profound anti-inflammaging effects by optimizing metabolic signaling, reducing ectopic adiposity, restoring $\beta$-cell function, and potentially facilitating mechanisms essential for telomere stability and cellular lifespan extension 4546.
Therapeutic Interventions: From Senolytics to Senomorphics
The translation of precision geroscience from bench to bedside hinges entirely on the development of highly targeted therapeutics designed to safely dismantle the molecular framework of inflammaging. These advanced interventions fall broadly into two primary categories: Senolytics (agents that selectively induce apoptosis in existing senescent cells) and Senomorphics (agents that suppress the SASP without killing the host cell).
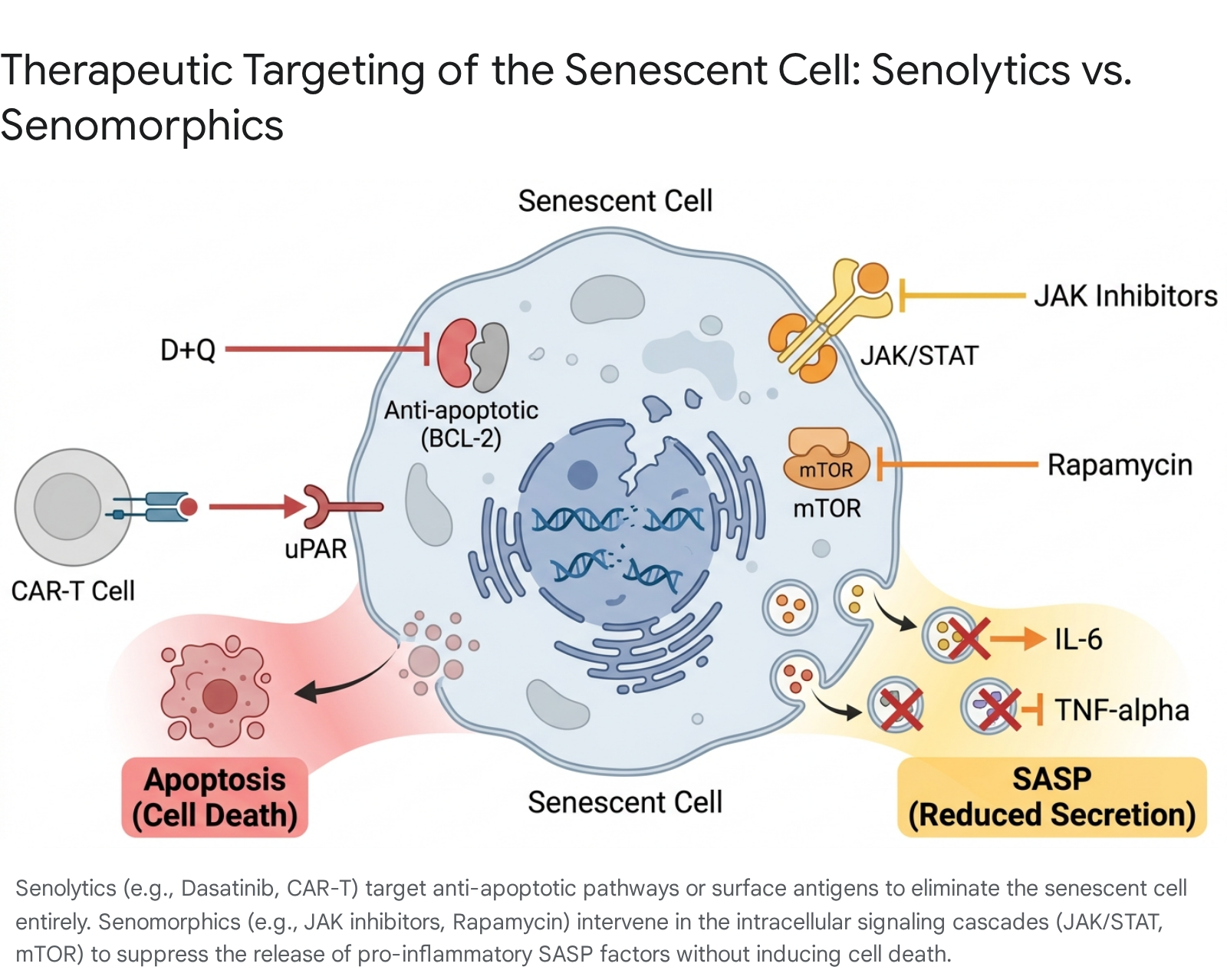
Table 2: Evaluation of Pharmacological Interventions Targeting Inflammaging
| Intervention Class | Specific Agents | Mechanism of Action | Efficacy & Clinical Readiness | Source |
|---|---|---|---|---|
| Senolytics | Dasatinib + Quercetin (D+Q) | Tyrosine kinase and flavonoid inhibitors targeting BCL-2 anti-apoptotic pathways to force elimination of senescent cells. | Experimental/Early Clinical: Shows clearance of senescence and gut dysbiosis reduction in mice. Phase I/II open-label trials (e.g., in early Alzheimer's) show CNS penetration, but broad clinical readiness is low due to systemic safety concerns. | 33344935 |
| Targeted Peptides | FOXO4-DRI | Specifically perturbs the FOXO4-p53 interaction in senescent cells, causing p53 nuclear exclusion and selective intrinsic apoptosis. | Preclinical: Restores tissue homeostasis, Leydig cell testosterone secretion, fur density, and renal function in aging mice. Clinical trials pending safety evaluations. | 3351 |
| Cellular Immunotherapy | in vivo CAR-T Cells (uPAR+) | Direct reprogramming of endogenous T-cells to target the urokinase plasminogen activator receptor (uPAR) on senescent cells using systemic LNP or viral vectors. | Preclinical/Translational: Highly effective in mice for lifespan extension and metabolic improvement. Translation is hindered by major limitations: vector immunogenicity, risk of insertional mutagenesis (with viral vectors), and severe toxicities like CRS and ICANS. | 343653 |
| Senomorphics (JAK Inhibitors) | Ruxolitinib, Baricitinib, Upadacitinib | Directly inhibits JAK/STAT intracellular signaling, actively blocking the downstream transcription of pro-inflammatory SASP cytokines (IL-6, IL-8). | Clinically Available (Repurposed): Highly effective in severe autoimmune diseases (e.g., Alopecia, Atopic Dermatitis, RA). However, systemic use carries FDA black-box warnings for major cardiovascular events (MACE), thrombosis, and serious infections, particularly in the frail elderly. Topical creams (Opzelura) show safer localized profiles. | 19353755383940 |
| Metabolic Modulators | Metformin, Rapamycin, GLP-1 Agonists | Modulates mTOR and AMPK pathways, enhancing autophagy, improving systemic insulin sensitivity, and reducing SASP translation. | Clinically Available: GLP-1 and Metformin have excellent safety profiles and are widely prescribed. Rapamycin requires careful, pulsed dosing to avoid immunosuppression. | 21463540 |
The Promise and Peril of Senolytics and Cellular Therapies
Senolytics like the Dasatinib and Quercetin (D+Q) combination have shown remarkable results in preclinical models, effectively reducing intestinal senescence, modulating the microbiome, and decreasing inflammatory markers 4935. In early-phase human trials focusing on older adults with early Alzheimer's disease, D+Q has demonstrated successful central nervous system (CNS) penetration and initial tolerability. Targeted transcriptomic analysis of peripheral blood mononuclear cells in these trials indicated a downregulation of inflammatory genes including FOS, FOSB, IL1$\beta$, and PTGS2 49. Advanced modalities, such as in vivo CAR-T cell therapies targeting uPAR-positive senescent cells, have successfully alleviated the systemic senescence burden, improved exercise capacity, and actively extended lifespan in murine models 3436.
However, the clinical readiness of systemic senolytics is frequently overstated in translational literature. Senescent cells play indispensable, highly regulated physiological roles in normal wound healing, tissue repair, and embryogenesis 33. The indiscriminate, systemic clearance of senescent cells carries severe theoretical risks of off-target toxicity and impaired tissue regeneration, leading to potential endocrine disruption 33. Furthermore, advanced in vivo CAR-T engineering faces immense translational hurdles. While Lipid Nanoparticle (LNP) delivery avoids some viral risks, the use of $\gamma$-retroviral or lentiviral vectors carries a severe risk of insertional mutagenesis and oncogene activation 3653. Additionally, these cellular therapies risk inducing severe adverse events, including massive Cytokine Release Syndrome (CRS) and Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS), which must be mitigated prior to broader clinical application 3653.
The Complex Calculus of JAK Inhibitors
Senomorphics, particularly Janus kinase (JAK) inhibitors like ruxolitinib, baricitinib, and upadacitinib, offer a drastically different approach by neutralizing the SASP without eliminating the host cell 3540. The JAK/STAT pathway is the primary signal transducer for multiple inflammaging-associated cytokines, including IL-6. In dermatology and rheumatology, JAK inhibitors have revolutionized the treatment of severe alopecia areata, atopic dermatitis, and rheumatoid arthritis 1937. The SELECT-COMPARE study demonstrated that upadacitinib outperforms adalimumab (a TNF inhibitor) in achieving sustained clinical remission in RA patients 19. Furthermore, topical formulations like ruxolitinib cream (evaluated in the TRuE-AD4 phase 3b trial) offer profound localized efficacy for atopic dermatitis without the risks associated with systemic exposure 38.
Yet, their generalized application as systemic anti-aging therapeutics is severely complicated by their safety profiles. The landmark ORAL Surveillance trial definitively established that in older adults (aged >50) with underlying cardiovascular risk factors, systemic pan-JAK inhibitors are associated with an elevated risk of major adverse cardiovascular events (MACE), venous thromboembolism, malignancies, and severe opportunistic infections compared to TNF inhibitors 1339. Consequently, the very demographic most fundamentally affected by inflammaging - the frail elderly - are often the poorest clinical candidates for systemic JAK inhibition due to their diminished physiological reserve 1339.
The Diagnostic Gap: The Missing Standardized Biomarker Panels
A critical bottleneck preventing the widespread clinical implementation of longevity interventions is the persistent "diagnostic gap." While chronologic age is easily measured, the precise quantification of biological age and the accurate stratification of patients based on their specific inflammaging burden requires robust, reproducible, and universally standardized biomarker panels 21594142. Currently, the lack of such stringent standardization severely hampers the design, reproducibility, and endpoint evaluation of senolytic clinical trials 4962.
In response to this critical gap, a recent, highly rigorous Delphi consensus process (spanning 2025 and 2026) involving leading international geroscience experts successfully identified 14 priority biomarkers of aging strictly designed for use as outcome measures in intervention studies 436444. This consensus panel spans multiple domains, identifying physiological markers like Insulin-like growth factor 1 (IGF-1) and DNA methylation-derived GDF15 (DNAmGDF15), the latter having emerged as an exceptionally robust marker linked to mitochondrial stress, sarcopenia, and epigenetic alterations 24364. It standardizes inflammatory markers, specifically highly sensitive C-reactive protein (hsCRP) and IL-6 4364. Functionally, the consensus relies on highly quantifiable metrics: muscle mass, hand grip strength (HGS), gait speed, the Timed-Up-and-Go (TUG) test, standing balance, the Frailty Phenotype (FP), cognitive health, and blood pressure 4364. Finally, it incorporates advanced epigenetic clocks to measure the DNA methylation pace of aging 4364.
The absolute necessity of these markers was demonstrated in the observational longitudinal BASE-II study, which tracked 1,083 older adults over 7.4 years. Extensive analysis comparing these 14 consensus biomarkers demonstrated that the epigenetic pace of aging (specifically DunedinPACE) alongside functional metrics like standing balance and muscle mass, emerged as the strongest and most consistent predictors of mortality, far outperforming single inflammatory cytokines alone 64.
Furthermore, advancements in assessing immune aging rely on specific T-cell metrics. By monitoring thymic output via SOX4/CD38 positive recent thymic emigrants (RTEs) and the expansion of GZMK+ CD8 cells, clinicians can accurately stage immune aging and benchmark rejuvenation strategies 34. Ultimately, the integration of spatial omics - a technology spearheaded by the NIH Cellular Senescence Network (SenNet) which allows researchers to map senescent cells within the precise spatial architecture of a tissue - will be essential for differentiating physiological, beneficial senescence from pathological inflammaging 62. Until these multidimensional, standardized biomarker panels undergo rigorous independent validation and achieve broad regulatory approval, the clinical administration of advanced senotherapeutics will remain largely experimental, confined to the realm of discovery rather than routine diagnostic medicine 4942.
Conclusion
Inflammaging represents the profound physiological convergence of immunosenescence, mitochondrial dysfunction, metabolic exhaustion, and environmental mismatch. As definitively elucidated by recent multi-population studies in non-industrialized cohorts, this chronic, sterile inflammation is not an inescapable consequence of the passage of time, but a highly modifiable biological trajectory dictated heavily by lifestyle, geographic, and socio-environmental contexts.
The successful translation of precision geroscience into clinical practice requires a nuanced, multi-modal approach. While behavioral interventions such as the rigorous optimization of slow-wave sleep and resistance-based exercise immunology provide immediate, highly effective, and risk-free physiological mechanisms to suppress systemic inflammation, pharmacological interventions demand significantly greater caution. The deployment of potent senolytics, engineered cellular therapies, and systemic JAK inhibitors holds profound theoretical promise but is currently heavily constrained by an unresolved diagnostic gap and the physiological complexities of the frail elderly. The path forward dictates an urgent, global prioritization of standardized, consensus-driven biomarker panels - integrating epigenetic clocks, dynamic SASP profiles, and functional metrics - to safely transition precision longevity medicine from compelling biological theory into standard, evidence-based clinical reality.