Oral microbiome and systemic aging
Global Epidemiology of Periodontal Disease
Disease Prevalence and Demographic Trends
Periodontal disease is a chronic, progressive inflammatory condition characterized by the destruction of the periodontium, including the alveolar bone, periodontal ligament, and gingival tissues. It is widely recognized as one of the most prevalent non-communicable diseases globally, acting as a primary cause of tooth loss and a significant contributor to diminished quality of life in adult populations 12. Data extracted from the Global Burden of Disease (GBD) 2021 study provides a comprehensive epidemiological landscape of the condition. In 2021, severe periodontitis affected approximately 1.07 billion individuals worldwide 34. The global age-standardized prevalence rate (ASPR) for severe periodontitis was estimated at 12,498.3 per 100,000 population 34.
Between 1990 and 2021, the absolute number of global cases rose by an unprecedented 91.54% 34. Decomposition analyses demonstrate that this massive expansion in disease volume was primarily driven by sweeping demographic shifts rather than an inherent increase in disease infectivity or etiology. Population growth accounted for 66.15% of the increase, while population aging contributed 30.84% 34. Because periodontal disease involves cumulative tissue destruction over decades, the burden is heavily concentrated in older cohorts. Global prevalence peaks prominently among individuals aged 50 to 59 years, reflecting the chronic nature of the disease and prolonged exposure to localized pathogenic biofilms, metabolic dysregulation, and environmental risk factors such as smoking 345. While incidence rates have remained relatively stable, the age-standardized rates of both prevalence and disability-adjusted life years (DALYs) have increased, indicating that global populations are living longer with the disease and its associated morbidities 16.
Socioeconomic Impact and Regional Disparities
The distribution of periodontal disease exhibits profound geographic and socioeconomic disparities, deeply intertwined with healthcare access, oral health literacy, and structural economic inequalities. According to analyses stratifying populations by the Socio-demographic Index (SDI) - a composite metric of income, educational attainment, and fertility rates - there is a highly significant negative correlation between SDI and the burden of periodontal disease 6. Low and low-middle SDI regions report age-standardized prevalence rates that consistently exceed the global average. Specifically, the low-middle SDI region reported the highest ASPR at 15,252.3 per 100,000 population, while the middle SDI region carried the highest absolute volume of cases, numbering approximately 350 million 34.
At the regional level, South Asia represents the heaviest geographic burden, contributing roughly 310 million cases and an ASPR of 17,566.1 per 100,000 4. The vast majority of the global increase in total cases over the last three decades was concentrated in specific rapidly developing nations; India and China collectively accounted for 48.22% of the global rise 34. Conversely, high SDI regions benefit from superior healthcare infrastructures and allocate a larger percentage of public health funding to preventive dental care, which translates to a lower overall ASPR 5. However, these high SDI nations are experiencing an upward pressure on disease prevalence driven by profound demographic aging and improved tooth retention rates in older populations, which prolongs the temporal window for periodontitis to develop 7.
The socioeconomic consequences of this epidemiological distribution are severe. The global burden of periodontitis accounts for an estimated 3.5 million years lived with disability annually and results in productivity losses exceeding $54 billion 7. When combined with the direct costs of dental care, the total economic burden of oral diseases - of which periodontitis is a primary component - reaches approximately $442 billion each year 7. Projections utilizing the Bayesian Age-Period-Cohort (BAPC) model extending to 2040 indicate a continued rise in total cases across all SDI regions, necessitating systemic policy reforms and the integration of oral health into universal health coverage to mitigate the escalating public health crisis 16.
| Epidemiological Metric (GBD 2021) | Global Estimate / Percentage | Key Drivers and Regional Concentrations |
|---|---|---|
| Total Global Prevalent Cases | ~1.07 Billion | South Asia, East Asia |
| Global ASPR (per 100,000) | 12,498.3 | Highest in Low-middle SDI Regions |
| Total Case Growth (1990-2021) | +91.54% | India and China (48.22% of total growth) |
| Growth Attribution: Population Growth | 66.15% | Dominant driver in Low SDI Regions |
| Growth Attribution: Population Aging | 30.84% | Dominant driver in High and Middle SDI Regions |
Pathobiology of the Oral-Gut-Systemic Axis
Pathogen Translocation and Endothelial Dysfunction
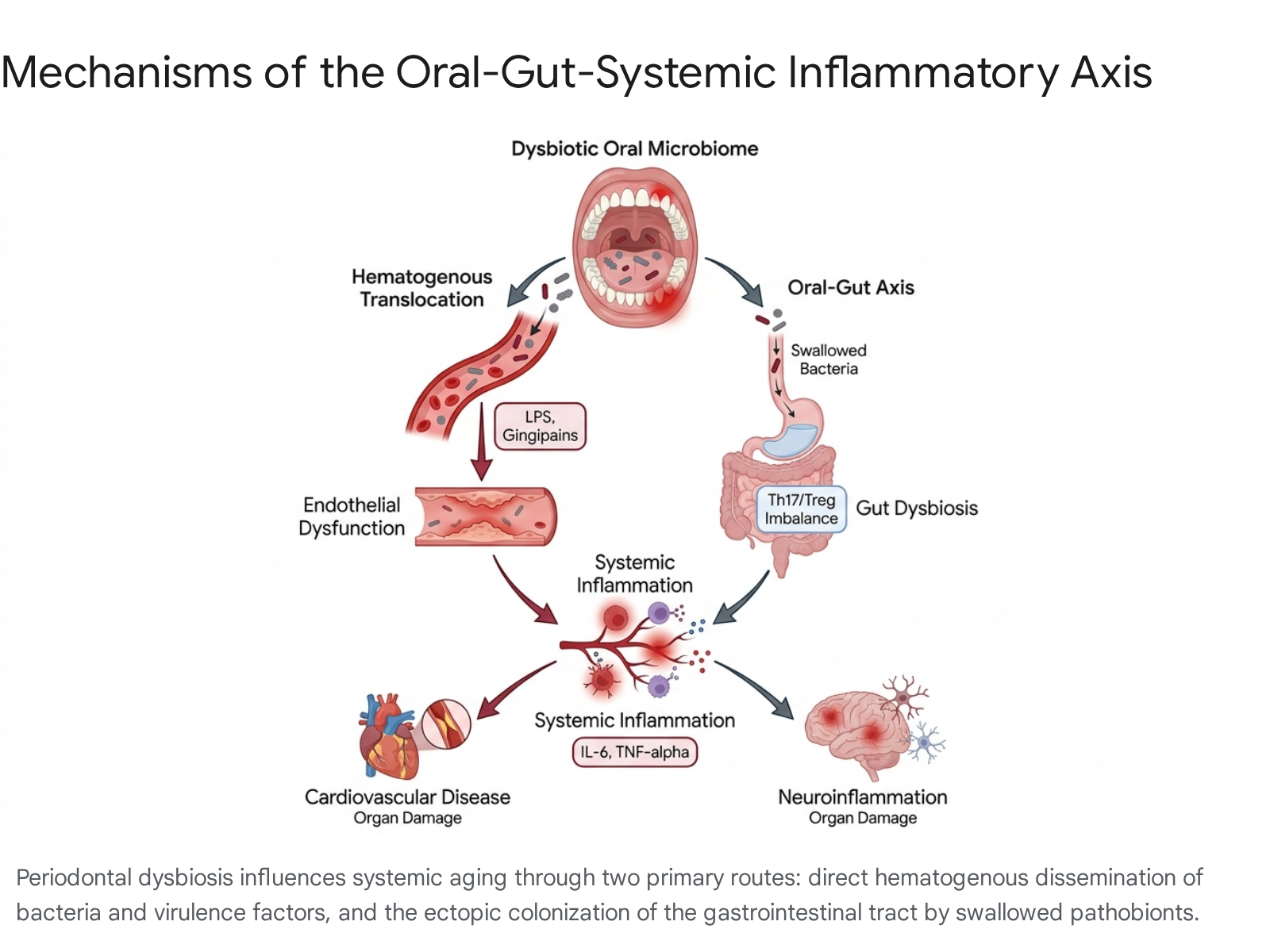
The oral cavity harbors a complex, diverse microbiome that, under homeostatic conditions, acts as a protective barrier preventing the colonization of exogenous pathogens and actively modulating the local mucosal immune system 89. However, poor oral hygiene, environmental stressors, and systemic metabolic changes can disrupt this equilibrium, leading to oral dysbiosis. This dysbiotic state is marked by a profound taxonomic shift: the depletion of health-associated, nitrate-reducing commensals (such as Neisseria, Rothia, and Haemophilus) and the rapid expansion of proteolytic anaerobic pathobionts, including Porphyromonas gingivalis, Fusobacterium nucleatum, and Prevotella intermedia 810.
This localized dysbiosis initiates a chronic inflammatory state that progressively degrades the integrity of the gingival epithelium. The resulting ulcerated periodontal pockets serve as a chronic portal for microbial dissemination into the host. Through routine physiological activities such as mastication and mechanical oral hygiene, bacteria and their associated virulence factors - namely lipopolysaccharides (LPS) and specialized proteolytic enzymes known as gingipains - gain direct access to the systemic circulation 81011. Hematogenous dissemination exposes distant organ systems and the vascular endothelium to sustained, low-grade microbial insults 91012.
Once in the bloodstream, pathogen-associated molecular patterns (PAMPs) such as LPS activate Toll-like receptors (specifically TLR2 and TLR4) and NOD-like receptors (NLRs) on the surface of endothelial and circulating immune cells 10. This binding initiates NF-κB-driven signaling cascades, resulting in a systemic immunometabolic signature defined by chronic low-grade cytokinemia. Elevated circulating levels of pro-inflammatory cytokines, specifically interleukin-1 beta (IL-1β), interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and C-reactive protein (CRP), drive widespread systemic inflammation 810. Quantitatively, systemic IL-6 levels can increase by 25 - 35%, and CRP by 1 - 3 mg/L in patients with severe periodontitis 10. Furthermore, P. gingivalis outer membrane vesicles are capable of penetrating endothelial monolayers, directly disrupting cellular tight junctions and increasing vascular permeability 10. This cascade fosters endothelial activation - characterized by the upregulation of adhesion molecules like ICAM-1 and VCAM-1 - which facilitates the recruitment of leukocytes into the vessel wall, a critical initiating step in atherogenesis and vascular aging 810.
The Oral-Gut Microbiome Interaction
Beyond hematogenous dissemination, emerging research identifies the oral-gut axis as a critical bidirectional pathway mediating systemic aging and disease 1314. Humans continuously swallow saliva, ingesting an estimated $10^9$ to $10^{10}$ bacterial cells daily 14. While gastric acidity serves as a primary physiological defense, periodontopathic bacteria such as P. gingivalis and F. nucleatum exhibit robust acid tolerance and routinely survive transit to reach the lower gastrointestinal tract 1415.
Upon successful ectopic colonization in the gut, these oral pathobionts severely disrupt the native microbial ecology, initiating a state of secondary intestinal dysbiosis 1415. P. gingivalis alters gut homeostasis by suppressing beneficial metabolic outputs, notably the production of short-chain fatty acids (SCFAs) like acetate, propionate, and butyrate, which are essential for maintaining colonocyte health and immune regulation 81314. The presence of F. nucleatum directly increases inflammatory gene expression through the binding of its FadA adhesin complex to E-cadherin receptors on intestinal epithelial cells, triggering downstream NF-κB activation 15. This disruption compromises the intestinal epithelial barrier, leading to increased intestinal permeability, commonly referred to as "leaky gut" 1315.
Immunological Imbalance and Th17/Treg Disruption
The immunological consequences of ectopic gut colonization are profound and systemic. Ectopic P. gingivalis drives the differentiation of pathobiont-reactive T-helper 17 (Th17) cells within the gut-associated lymphoid tissue (GALT) 16. Normally, immune homeostasis in the gut is maintained by a delicate balance between pro-inflammatory Th17 cells and immunosuppressive Regulatory T cells (Tregs). P. gingivalis actively disrupts this balance by altering tryptophan metabolism in the gut, reducing beneficial aryl hydrocarbon receptor (AHR) ligands, and increasing toxic metabolites such as 3-indoleacrylic acid and N'-formylkynurenine, which are linked to neutrophil activation and neurotoxicity, respectively 1314. It also suppresses the production of gut-protective mediators like resolvin D5n-3 DPA, further exacerbating the Th17/Treg imbalance 13.
These induced Th17 cells, alongside locally produced pro-inflammatory cytokines, translocate into the systemic circulation, causing widespread immunological maladaptation 141516. Moreover, the compromised gut barrier allows intestinal-derived endotoxins to enter the bloodstream, resulting in metabolic endotoxemia 1015. This dual assault from hematogenous oral pathogens and translocated gut endotoxins synergistically amplifies systemic inflammation, accelerating endothelial degradation, metabolic dysregulation, and neuroinflammation 101516.
Dietary Modulation of Oral Ecology and Aging
Impact of High-Sugar and Processed Diets
Systemic aging and oral microbial ecology are significantly modulated by overarching dietary patterns. High and frequent intake of refined sugars disrupts localized metabolic homeostasis in the oral cavity, fueling the proliferation of acidogenic and periodontopathic bacteria 171819. Sugar-rich diets heavily select for pathogenic organisms such as Streptococcus mutans and Porphyromonas gingivalis, establishing an ecological niche permissive to both dental caries and periodontitis 1719. Beyond the local oral environment, excessive sugar consumption contributes to systemic insulin resistance, elevated oxidative stress, and chronic low-grade inflammation, serving as an accelerator for cognitive decline and metabolic dysfunction over the lifespan 1718.
The Nitrate-Nitrite-Nitric Oxide Pathway
Conversely, diets rich in inorganic nitrates - such as those abundant in leafy green vegetables like spinach, arugula, and beetroot - exert a highly protective, prebiotic effect on the oral microbiome and subsequent systemic health 1720. Dietary nitrate follows a unique metabolic trajectory: after gastrointestinal absorption, approximately 25% of circulating nitrate is actively concentrated in the salivary glands and secreted back into the oral cavity 1821.
Within the oral environment, facultative anaerobic commensal bacteria, notably Rothia and Neisseria, utilize nitrate reductases to convert this salivary nitrate into nitrite 182021. This microbially produced nitrite is subsequently swallowed, entering the highly acidic environment of the stomach and the systemic circulation, where it undergoes a secondary reduction to form nitric oxide (NO) 2021.
Nitric oxide is a potent, short-lived signaling molecule and vasodilator that is absolutely crucial for regulating systemic blood pressure, maintaining vascular endothelial homeostasis, and facilitating efficient cerebral blood flow and neurovascular coupling 2021. Biological aging naturally diminishes endogenous NO production via classical enzymatic pathways, making the microbiome-dependent nitrate-nitrite-NO pathway increasingly critical for cardiovascular and cognitive maintenance in older adults 2021. Research confirms that nitrate-rich diets actively increase the relative abundance of health-associated oral taxa while concurrently reducing populations of disease-associated genera like Prevotella and Veillonella 1821. By optimizing systemic nitric oxide bioavailability, these microbiome-mediated dietary interventions mitigate endothelial dysfunction, reduce arterial stiffness, and exhibit significant protective effects against age-related neurodegeneration 171820.
| Dietary Exposure | Primary Effect on Oral Microbiome | Downstream Systemic and Aging Outcomes |
|---|---|---|
| High Refined Sugar | Proliferation of S. mutans, P. gingivalis; lowered pH. | Promotes metabolic endotoxemia, insulin resistance, accelerated cognitive decline, and systemic inflammation. |
| High Inorganic Nitrate | Expansion of nitrate-reducing commensals (Rothia, Neisseria). | Elevates systemic Nitric Oxide (NO), improves endothelial vasodilation, lowers blood pressure, and enhances cerebral blood flow. |
| High Dietary Fiber | Increased microbial diversity; SCFA production. | Supports intestinal barrier integrity, reduces systemic oxidative stress, and promotes immune regulation (Treg balance). |
Epigenetic Clocks and Biological Aging Acceleration
Fundamentals of Epigenetic Aging Markers
The chronic inflammatory burden generated by oral dysbiosis leaves quantifiable and lasting marks on the host's molecular machinery, particularly through epigenetic modifications. The distinction between chronological age and biological age is central to modern geroscience. While chronological age measures the passage of time, biological age captures the accumulated molecular, physiological, and environmental damage that ultimately dictates frailty and disease vulnerability 222324.
Epigenetic clocks - complex machine-learning algorithms based on specific DNA methylation patterns at short DNA regions rich in cytosine-guanine dinucleotides (CpG islands) - are currently the most accurate biomarkers for assessing biological aging 2324. First-generation clocks primarily estimated chronological age, whereas second-generation clocks are designed to predict physiological decline, mortality, and age-related disease risk. For example, the GrimAge and Hannum clocks focus heavily on mortality prediction and epigenetic responses to lifestyle factors, while DNAm PhenoAge integrates clinical biomarkers (such as glucose and inflammatory markers) with methylation data to capture a broader, highly disease-sensitive spectrum of physiological aging 222425. Discrepancies where epigenetic age exceeds chronological age are termed "epigenetic age acceleration" and strongly correlate with chronic health conditions 2225.
Causality Between Periodontitis and Phenotypic Age
Recent genetic analyses have investigated the direct causal links between periodontal inflammation and accelerated biological aging. In a large-scale Mendelian randomization (MR) study exploring the causality of periodontitis on various age acceleration measures, researchers identified a significant genetic causal relationship specifically between periodontitis and DNAm PhenoAge acceleration (Inverse Variance-Weighted [IVW] β=0.308; P=0.017) 22. While the study found no causal link to other metrics like GrimAge or intrinsic epigenetic age acceleration (IEAA), the specific impact on PhenoAge suggests that periodontitis fundamentally drives the systemic inflammatory components of the aging phenotype 22.
This biological aging acceleration is mediated through shared pleiotropy. Statistical analyses revealed 24 shared SNPs associated with 242 genes that overlap between periodontitis and accelerated aging, predominantly involving immune functions and cellular senescence pathways 22. Further eQTL integration analysis highlighted 91 genes causally linked to both conditions, identifying specific loci such as C6orf183 as potential mechanistic mediators 22. The continuous host-pathogen interaction in the periodontium induces profound DNA methylation and histone modifications that govern the expression of key pro-inflammatory cytokines (IL-1, IL-6, TNF-α) 262728. Chronic systemic exposure to these cytokines accelerates "inflammaging" - the progressive deterioration of the immune system and the gradual accumulation of sterile, low-grade systemic inflammation over the lifespan 29. Consequently, successful clinical management of periodontitis may possess the therapeutic potential to decelerate epigenetic aging and extend human healthspan 222330.
Periodontitis and Cardiovascular Health
Observational Evidence and Mortality Risk
Extensive observational and epidemiological data have historically demonstrated a robust association between periodontal disease and elevated cardiovascular morbidity and mortality. Large-scale meta-analyses encompassing millions of clinical participants report that individuals with severe periodontitis experience a significantly elevated risk of all-cause mortality, with a hazard ratio (HR) of 1.46, and cardiovascular mortality, with an HR of 1.47 31. Furthermore, periodontitis is clinically associated with substantially increased risks for incident coronary heart disease, myocardial infarction, and cerebrovascular diseases, including ischemic stroke (HR 2.20) 23132.
The mechanistic rationale supporting these observational findings aligns closely with the endothelial dysfunction models previously discussed. Periodontal pathogens and systemic inflammatory mediators promote vascular rigidity, atherosclerotic plaque formation, and prothrombotic signaling, directly injuring the cardiac endothelium 21233. Longitudinal cohort studies confirm that individuals suffering simultaneously from severe periodontitis and high baseline systemic inflammation face a compounded, synergistic risk, manifesting a 28% higher likelihood of mortality from cardiovascular disease compared to healthy controls 11. However, assessing true biological causality in observational research is inherently complex due to ubiquitous confounding variables. Risk factors such as smoking, lower socioeconomic status, and poor dietary habits are highly prevalent in populations with both periodontitis and cardiovascular disease. These shared lifestyle factors routinely inflate observational hazard ratios, creating a "Janus effect" where statistical models yield wildly different associations depending on which covariates are adjusted for 313435.
Mendelian Randomization and Genetic Causality
To circumvent the limitations of residual confounding and reverse causality inherent in observational cohort studies, researchers increasingly utilize Mendelian Randomization (MR). MR functions similarly to a randomized controlled trial by utilizing genetic variants - specifically single nucleotide polymorphisms (SNPs) - as instrumental variables to infer direct causality. Because these alleles are randomly assorted at conception, their distribution is generally independent of environmental and lifestyle confounders 363738.
Recent comprehensive MR analyses have challenged several long-held clinical assumptions regarding the direct causal effects of periodontitis on the cardiovascular system 394041. For decades, clinical practice mandated prophylactic antibiotic use prior to dental procedures to prevent infective endocarditis (IE), based on strong observational links. However, rigorous MR studies utilizing large-scale GWAS data found absolutely no genetic causal association between acute, chronic, or aggressive periodontitis and infective endocarditis (OR 0.947 for chronic periodontitis) 4243. Similarly, bidirectional MR analyses investigating coronary atherosclerosis, heart failure, and overall ischemic stroke found no significant causal link originating from periodontitis (OR 1.00 for coronary atherosclerosis) 39404143.
| Systemic Outcome Evaluated | Mendelian Randomization Result (Causal Effect) | Evidence Strength and Odds Ratio (OR) |
|---|---|---|
| Cardioembolic Stroke | Significant Positive Causal Link | Strong evidence; OR 1.03 (95% CI 1.00-1.05) 41 |
| Major Depression | Significant Positive Causal Link | Strong evidence indicating increased risk 39 |
| Infective Endocarditis | No Causal Link | Robustly negative; OR 0.947 (Chronic) 4243 |
| Coronary Atherosclerosis | No Causal Link | Robustly negative; OR 1.00 40 |
| Ischemic Stroke (Overall) | No Causal Link | Robustly negative; OR 1.00 41 |
| Alzheimer's Disease | No Causal Link | Robustly negative; OR ~0.97 37 |
These null findings suggest that the profound correlations observed in epidemiological studies are heavily mediated by shared genetic risk factors, the overarching systemic inflammaging phenotype, and powerful lifestyle confounders, rather than periodontitis acting as a direct cardiovascular pathogen 313439.
Influence on Systemic Immune Cell Phenotypes
While periodontitis may not be the direct genomic catalyst for conditions like coronary atherosclerosis, it actively shapes a pro-inflammatory systemic environment that exacerbates widespread vascular degradation. Bidirectional MR analysis verifies that periodontitis causally modulates circulating immune cell phenotypes. Genetically predicted periodontitis drives the proliferation of T-helper 17 (Th17) cell populations, alters conventional dendritic cell (cDC) maturation, and elevates circulating chemokines such as CXCL11 and protein S100A12 444545. Conversely, it impacts protective immune regulatory proteins, identifying markers like PD-L1 and eotaxin as having a protective role against periodontal breakdown 44. This confirms that periodontitis is an active participant in systemic immune priming, fueling the chronic low-grade inflammation necessary for cardiovascular events to occur.
Neurodegeneration and Cognitive Decline
The Pathogen Hypothesis of Alzheimer's Disease
The intersection between oral health, microbial translocation, and cognitive decline represents one of the most vigorously debated areas of contemporary neurodegenerative research. The traditional view of Alzheimer's disease (AD) pathogenesis centers on the spontaneous accumulation of amyloid-beta (Aβ) plaques and tau neurofibrillary tangles. However, the emerging "pathogen hypothesis" posits that these accumulations are, in part, aggressive innate immune responses to microbial infiltration in the brain 364647. Evidence from in vitro and in vivo studies demonstrates that Aβ oligomers possess potent, broad-spectrum antimicrobial properties; they form dense fibrils that entrap translocated pathogens and disrupt their membranes, suggesting that amyloid plaques may initially serve a protective role before the inflammatory response loses regulation 4647.
Porphyromonas gingivalis has been positioned as a central infectious agent in this hypothesis. Observational and post-mortem tissue analyses have repeatedly detected P. gingivalis DNA and its toxic secreted proteases, known as gingipains, in the brain parenchyma and cerebrospinal fluid of AD patients at significantly higher rates than age-matched healthy controls 4849. Gingipains are specialized cysteine proteinases that cleave human host proteins, effectively degrading the neuronal cytoskeleton, destroying synapses, and actively promoting tau hyperphosphorylation 3348. In murine models, chronic oral infection with P. gingivalis results in the successful infiltration of the bacteria into the brain, robust microglial activation, subsequent Aβ accumulation, and severe hippocampal neurodegeneration 4849.
Clinical Trials of Gingipain Inhibitors
Based on this compelling preclinical and post-mortem evidence, the pharmaceutical industry aggressively pursued targeted gingipain inhibition as a potential first-in-class, disease-modifying therapy for Alzheimer's disease. The therapeutic candidate Atuzaginstat (COR388) was developed as an orally bioavailable, highly blood-brain barrier penetrant small molecule designed to irreversibly bind and deactivate lysine gingipains 464849. Because P. gingivalis uniquely resides intracellularly within host cell vacuoles and feeds exclusively on host proteins rather than carbohydrates, standard broad-spectrum antibiotics are highly ineffective at eradicating it from neural tissue; blocking gingipains chemically deprives the bacteria of its food supply and halts targeted tissue destruction 4850.
The investigational drug rapidly progressed to the Phase II/III GAIN trial (NCT03823404), a large, double-blind, placebo-controlled study involving 643 participants diagnosed with mild-to-moderate Alzheimer's disease 495152. Participants were randomized to receive 40 mg or 80 mg of atuzaginstat, or a matching placebo, twice daily for a duration of 48 weeks. Ultimately, the trial failed to meet statistical significance for its co-primary cognitive and functional endpoints (the ADAS-Cog11 and ADCS-ADL scoring scales) across the overall patient cohort 495152.
However, pre-specified subgroup analyses yielded highly intriguing data regarding the mechanism of action. Among the specific subgroup of 242 participants who had detectable P. gingivalis DNA in their saliva at the baseline screening, the 80 mg dose of atuzaginstat was associated with a statistically significant 57% slowing of cognitive decline on the ADAS-Cog11 scale, and reductions in salivary P. gingivalis loads strongly correlated with improved clinical outcomes 495152. Despite verifying target engagement by successfully reducing gingipain activity and modifying downstream AD biomarkers, the drug exhibited severe safety profile failures, specifically dose-related hepatotoxicity 5152. Approximately 15% of patients in the 80 mg arm developed liver enzyme elevations exceeding three times the upper limit of normal, alongside bilirubin elevations 4952. Furthermore, the dropout rate for the treatment group was exceptionally high at 40%, compared to 25% in the placebo group. This toxicity profile prompted the FDA to place a full clinical hold on atuzaginstat in January 2022, leading to the complete discontinuation of the program and a strategic pivot by the sponsor to next-generation compounds (COR588) and alternative disease targets 4951.
Reverse Causality and Bidirectional Mechanisms
The clinical failure of atuzaginstat in the broader Alzheimer's population prompted a rigorous reassessment of the causal architecture connecting periodontitis to dementia. Subsequent Mendelian randomization studies utilizing massive datasets from the FinnGen, IEU, and PGC databases evaluated the genetic causality between the two diseases 3637. Across multiple robust analyses - including Inverse Variance Weighting, weighted median, and MR-Egger regression - researchers found no convincing evidence to support periodontitis as a genetic causal factor in the development of Alzheimer's disease 363739.
The divergence between the strong observational presence of P. gingivalis in AD brains and the lack of genetic causality suggests a complex, bidirectional relationship heavily intertwined with aging, immune senescence, and functional decline 5354. Rather than acting as the primary singular etiological trigger, periodontitis is increasingly viewed as an opportunistic accelerator in neurodegeneration. A phenomenon of "reverse causality" is frequently observed: the progressive onset of cognitive impairment severely hampers a patient's ability to maintain adequate oral hygiene or comply with dental care. This neglect leads to rampant, unmanaged periodontal disease, which in turn fuels massive localized bacterial proliferation 5354. The resulting systemic inflammatory fires, driven by circulating cytokines and endotoxemia, subsequently exacerbate and accelerate existing neurodegeneration 5354. Furthermore, shared physiological mechanisms such as age-related blood-brain barrier degradation, systemic immune activation, and overall frailty create a devastating feedback loop where systemic aging and oral dysbiosis mutually reinforce one another, accelerating the trajectory toward mortality 1055.