Neuroinflammation in Depression Anxiety and Cognitive Decline
Introduction to the Neuroimmune Paradigm
For the better part of a century, the pathophysiological frameworks governing psychiatric and cognitive disorders were dominated by neurotransmitter-centric models. Conditions such as major depressive disorder (MDD), generalized anxiety disorder (GAD), and Alzheimer's disease were largely conceptualized as discrete malfunctions of monoamine synthesis or localized protein aggregation within the central nervous system. However, modern scientific consensus has increasingly adopted a neuroimmunological framework, recognizing that the brain is in continuous, dynamic communication with the systemic immune system. This paradigm shift posits that immune processes in the brain - specifically neuroinflammation - serve as a central etiology and progression driver for numerous psychiatric and neurodegenerative conditions 12.
The concept of neuro-immunology, brought to mainstream scientific prominence by publications such as Ed Bullmore's "The Inflamed Mind," refutes the historical notion of the brain as a strictly immune-privileged organ. Instead, contemporary literature positions systemic inflammation as a precursor and catalyst for central nervous system dysfunction 13. Emerging evidence indicates that peripheral inflammatory markers can predict the onset of psychiatric illness decades before clinical manifestation 45. This has led to the operationalization of an "inflammatory biotype," a substantial subgroup of patients whose psychiatric or cognitive symptoms are fundamentally driven by hyperactive immune signaling rather than primary neurochemical deficits 267.
Understanding the complex etiology of neuroinflammation requires a detailed examination of how peripheral immune signals breach the central nervous system, how resident glial cells translate these signals into neurotoxicity, and how environmental factors continually feed this systemic inflammatory loop. Identifying these mechanisms is critical, as it transitions the field toward precision psychiatry, wherein immunomodulatory interventions can be prescribed to patients exhibiting specific inflammatory biomarker profiles.
Pathways of Peripheral Immune Signaling to the Brain
The translation of peripheral systemic inflammation into central neuroinflammation does not occur through a single vector. The central nervous system utilizes highly specialized, multifaceted communication pathways to monitor systemic immune status.
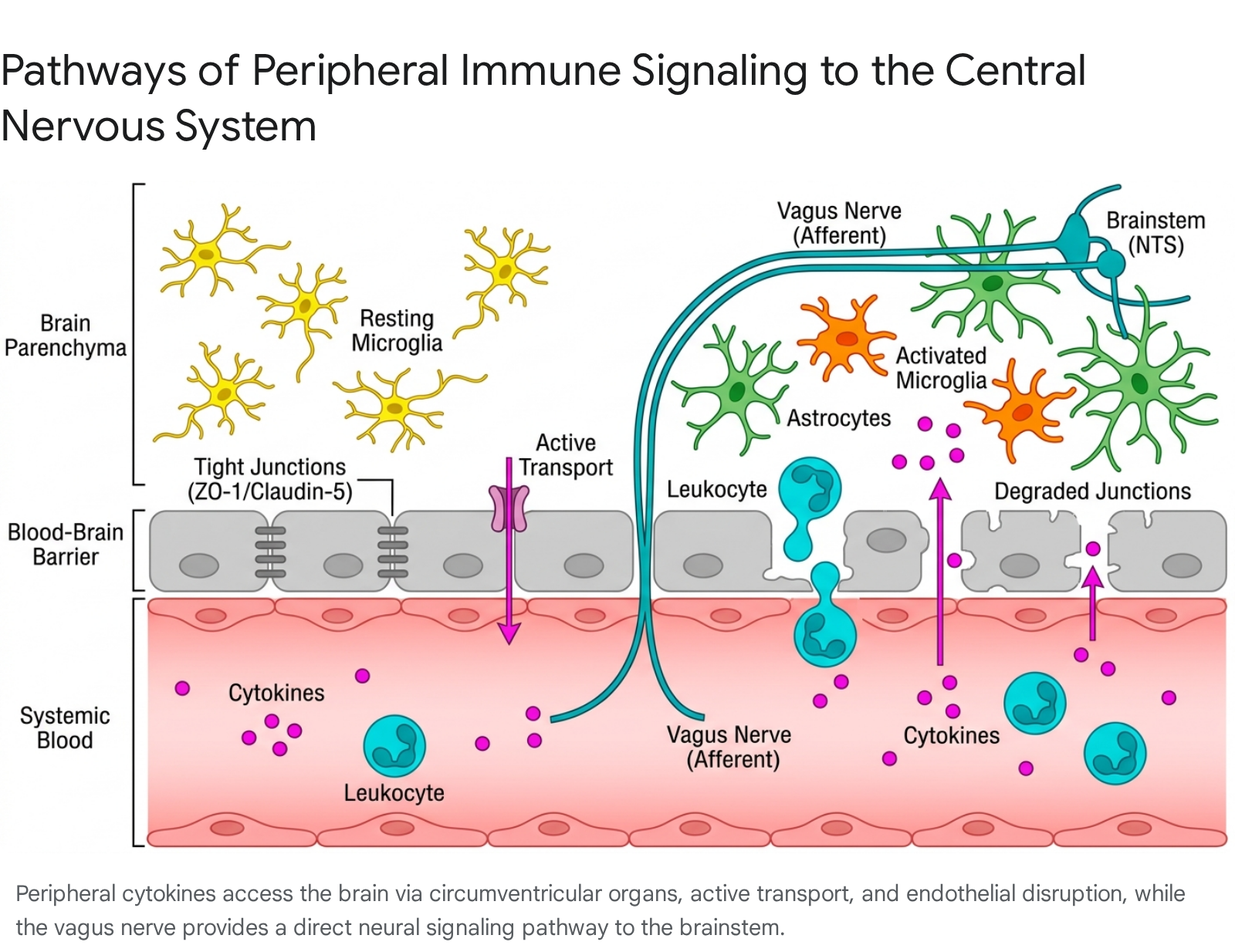
When peripheral inflammation is acute or chronically elevated, these physiological surveillance mechanisms become conduits for pathology.
The Blood-Brain Barrier and Endothelial Disruption
Under physiological conditions, the blood-brain barrier (BBB) functions as a highly selective diffusion barrier between the peripheral circulation and the central nervous system. The BBB is comprised of specialized brain microvascular endothelial cells (BMECs) linked by complex tight junctions and adherens junctions, supported by a basement membrane, pericytes, and astrocytic end-feet 8. However, severe or sustained peripheral inflammation triggers conditions that cause the BBB to lose its restrictive properties.
Systemic cytokines - predominantly tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6) - can bypass the BBB through circumventricular organs (CVOs), which naturally lack continuous endothelial tight junctions 910. Additionally, circulating cytokines can cross the intact BBB via saturable transport systems and receptor-mediated endocytosis 810. When systemic inflammation reaches critical thresholds, it induces a reactive response in the cerebral vascular endothelium itself. Pro-inflammatory molecules stimulate endothelial cells to express adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), which facilitate the tethering and rolling of peripheral leukocytes 1112.
Humoral and Cellular Infiltration Mechanisms
The humoral route of neuroinflammation describes the process by which systemic cytokine cascades directly damage the endothelial architecture. TNF-α and IL-1β trigger the dynamic reorganization of the actin cytoskeleton and the downregulation of essential tight junction proteins, including zonula occludens-1 (ZO-1), claudin-5, and occludin 1213. In vitro co-culture models demonstrating the interaction between lipopolysaccharide-activated microglia and brain endothelial cells show that this cytokine storm leads to a measurable decrease in trans-endothelial electrical resistance (TEER), indicating the collapse of barrier integrity 121314.
This structural degradation facilitates the cellular route of neuroinflammation. Paracellular gaps allow peripheral immune cells, such as monocytes, macrophages, and T-helper cells (Th1 and Th17), to infiltrate the brain parenchyma 1415. Once inside the central nervous system, these infiltrating cells cooperate with brain-resident microglia to release further cytokines and reactive oxygen species (ROS), amplifying BBB damage and driving synaptic dysfunction 1215.
Vagal Nerve and Autonomic Signaling Conduits
Beyond the vascular interface, the peripheral immune system communicates directly with the brain via rapid neural conduits, predominantly the vagus nerve (Cranial Nerve X). The vagus nerve operates as a sensory highway, detecting inflammatory states in visceral organs such as the gut and lungs and transmitting this information to the brainstem 161718.
Sensory afferent neurons within the nodose ganglia of the vagus nerve are equipped with cytokine receptors. Specifically, peripheral elevations in TNF-α and IL-1β sensitize transient receptor potential vanilloid 1 (TRPV1) channels on these neurons 1819. Upon activation, vagal afferents relay signals to the nucleus tractus solitarius (NTS) in the brainstem 1720. Glutamatergic projections from the NTS subsequently transmit these inflammatory signals to forebrain regions, including the paraventricular nucleus (PVN) of the hypothalamus and the bed nucleus of the stria terminalis (BNST) 1719.
This neural circuitry is responsible for generating rapid behavioral adaptations to illness, commonly referred to as "sickness behavior," which shares significant phenotypic overlap with major depressive disorder, including anhedonia, lethargy, and social withdrawal. However, chronic peripheral inflammation - such as that caused by colitis or acute pancreatitis - can persistently scramble these neuro-immune signals, disrupting the brain's homeostatic control over the immune response and embedding depressive and anxious symptomatology into the neural circuitry 1619.
Cellular Mediators: Microglia and Astrocytes
Once peripheral inflammatory signals successfully penetrate the central nervous system, they are processed and amplified by the brain's resident glial cells. The dynamic crosstalk between microglia and astrocytes is the primary cellular engine driving neuroinflammation, psychiatric symptoms, and cognitive decline.
Microglial Activation and the NLRP3 Inflammasome
Microglia are the principal innate immune cells of the central nervous system. In a healthy physiological state, they exist in a "ramified" resting morphology, actively surveilling the local microenvironment for damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) 1021. They also perform vital maintenance tasks, including synaptic pruning and the clearance of cellular debris. However, upon exposure to translocated peripheral cytokines, oxidative stress, or BBB leakage, microglia undergo morphological and functional transformations into an "amoeboid," reactive, pro-inflammatory phenotype 1222.
A critical mechanism of this microglial reactivity involves the Nod-like receptor protein 3 (NLRP3) inflammasome. The NLRP3 complex is highly responsive to stress-related mitochondrial dysfunction and reactive oxygen species 23. Upon activation, the inflammasome facilitates the cleavage and maturation of pro-inflammatory interleukins, specifically IL-1β and IL-18, releasing them into the neural parenchyma 723. This localized cytokine release directly alters neurotransmitter metabolism. Pro-inflammatory cytokines upregulate the enzyme indoleamine 2,3-dioxygenase (IDO), which alters tryptophan metabolism. Instead of synthesizing serotonin, tryptophan is shunted down the kynurenine pathway, resulting in the production of neurotoxic metabolites such as quinolinic acid. This biochemical diversion links microglial activation directly to the depletion of mood-regulating neurotransmitters and the onset of depressive symptoms 79.
Astrocytic Reactivity and Neurotoxic Cascades
Microglia do not drive neuroinflammation in isolation; their signaling profoundly dictates the behavior of astrocytes. Astrocytes are the most abundant glial cells in the brain, responsible for maintaining the BBB, regulating cerebral blood flow, and providing trophic support to neurons 1224. Recent pathophysiological studies have mapped a direct signaling axis where activated microglia secrete a specific cocktail of molecules - comprising IL-1α, TNF-α, and complement component C1q - that induces astrocytes to transition into a highly reactive, neurotoxic "A1" state 24.
Once in the A1 state, astrocytes lose their neuro-supportive functions. They cease promoting synaptogenesis and fail to phagocytose myelin debris. Instead, they secrete potent, though not yet fully identified, neurotoxins that induce rapid apoptosis in neurons and oligodendrocytes 24. This microglia-astrocyte interaction is particularly prominent in the pathogenesis of Alzheimer's disease (AD) and other dementias. Positron emission tomography (PET) imaging studies utilizing radiotracers for translocator protein (TSPO) - a marker for glial activation - demonstrate that amyloid-beta (Aβ) pathology only correlates with severe astrocyte reactivity and subsequent cognitive decline when concurrent microglial activation is present 25. This indicates that protein aggregation alone is insufficient to cause dementia; it is the dysregulated neuro-immune response that ultimately destroys cognitive architecture 2526.
Table 1: Cellular Roles in the Neuroinflammatory Cascade
| Cell Type | Homeostatic Function | Reactive / Pathological Phenotype | Mediators Released | Clinical Implications |
|---|---|---|---|---|
| Endothelial Cells | Maintain BBB integrity and selective permeability. | Upregulate adhesion molecules (ICAM-1, VCAM-1); lose tight junctions. | Chemokines, Thrombin | Facilitates leukocyte infiltration; early step in sub-acute neuroinflammation. |
| Microglia | Synaptic pruning, immune surveillance, debris clearance. | Amoeboid morphology; NLRP3 inflammasome activation; halts pruning. | IL-1β, IL-6, TNF-α, ROS, C1q | Drives kynurenine pathway (serotonin depletion); initiates A1 astrocyte conversion. |
| Astrocytes | Trophic neuronal support, nutrient transport, BBB regulation. | "A1" Neurotoxic state; loss of phagocytic and supportive abilities. | Unknown neurotoxins, inflammatory chemokines | Direct execution of neuronal and oligodendrocyte death; cognitive decline. |
The "Inflammatory Biotype" in Psychiatric Populations
The recognition of neuroinflammation has led to the identification of an "inflammatory biotype," a clinically distinct subgroup of patients exhibiting specific physiological and symptomatic profiles across multiple psychiatric diagnoses. This biotype challenges the monolithic classification of mental health disorders, explaining why a significant percentage of patients fail to respond to standard monoaminergic therapies.
Inflammatory Markers in Major Depressive Disorder and Anxiety
Systematic reviews and meta-analyses consistently confirm that major depressive disorder is associated with low-grade systemic inflammation, characterized by elevated peripheral levels of C-reactive protein (CRP), IL-6, and TNF-α 227. A highly replicated clinical threshold establishes that patients with hs-CRP levels greater than 3 mg/L represent a discrete inflammatory depression subgroup. This subgroup presents with distinct clinical features, including profound anhedonia, severe psychomotor slowing, heightened cognitive impairment, and marked resistance to traditional selective serotonin reuptake inhibitors (SSRIs) 2.
The predictive validity of these biomarkers is robust. Data from the massive Swedish AMORIS cohort, which tracked over 585,000 individuals for up to 30 years, revealed that individuals who subsequently developed psychiatric disorders exhibited persistently elevated levels of leukocytes and haptoglobin, alongside diminished levels of anti-inflammatory immunoglobulin G (IgG), decades prior to diagnosis 45. Longitudinal structural equation modeling further indicates that elevated systemic inflammation acts as a central mechanism mediating the relationship between baseline MDD or generalized anxiety disorder (GAD) and severe decrements in executive function up to 18 years later 2829.
Furthermore, specific cytokine profiles correlate with distinct clinical presentations. Melancholic depression exhibits a strong positive correlation with elevated IL-6 and dysregulated cortisol. In contrast, atypical depression - characterized by symptom reversal such as hyperphagia and hypersomnia - is more robustly associated with elevated CRP levels 30.
Schizophrenia, Bipolar Disorder, and Transdiagnostic Features
The inflammatory biotype extends beyond unipolar depression into severe mental illnesses such as bipolar disorder and schizophrenia. In bipolar disorder, markers like CRP and TNF-α surge during acute manic or depressive episodes and often normalize during euthymia, indicating a state-dependent inflammatory response 2. However, IL-6 frequently remains persistently elevated regardless of mood state, suggesting an underlying chronic immune vulnerability 2.
In schizophrenia, elevated inflammatory markers correlate strongly with generalized cognitive impairments and altered neurobehavioral functions 31. Proteomic analyses reveal an upregulation of pro-inflammatory cytokines and a decrease in anti-inflammatory markers such as vitamin C, niacin, and folate among patients 32. It is crucial to note, however, that calibrated uncertainty remains in the literature. Inflammation is not a universal feature of schizophrenia; postmortem studies show that markers like IL-1β are inconsistently elevated across the patient population, reinforcing the concept that neuroinflammation defines a specific biotype rather than the entirety of the disorder 2331.
Systemic and Environmental Drivers of the Inflammatory Biotype
If chronic low-grade inflammation drives the pathophysiology of the inflammatory biotype, the origins of this peripheral immune activation must be identified. Emerging literature highlights the exposome - the totality of dietary, microbial, and environmental exposures - as the primary instigator of systemic immune dysregulation.
The Gut-Microbiome-Brain Axis
The human gastrointestinal tract hosts trillions of microorganisms that produce metabolic byproducts capable of interacting with the host's nervous and immune systems. Imbalances in this microbial community, known as dysbiosis, significantly impact neuroinflammation. Disruptions in the gut microbiota compromise the intestinal epithelial barrier, colloquially known as "leaky gut," allowing lipopolysaccharides (LPS) and other bacterial endotoxins to enter the systemic circulation 3334. This endotoxemia provokes a sustained peripheral immune response that propagates to the brain via the vagus nerve and circulating cytokines 1835.
Conversely, a balanced microbiome provides substantial neuroprotection. Beneficial microbial species, such as Bifidobacterium longum and Lactobacillus rhamnosus, ferment non-digestible dietary fibers into short-chain fatty acids (SCFAs), including butyrate, acetate, and propionate 3335. SCFAs are potent anti-inflammatory agents; they fortify blood-brain barrier integrity and modulate microglial maturation 3435. Furthermore, butyrate acts as a histone deacetylase (HDAC) inhibitor, utilizing epigenetic mechanisms to suppress pro-inflammatory gene expression and promote hippocampal neurogenesis 36. The depletion of SCFA-producing bacteria, frequently observed in cohorts with major depressive disorder and schizophrenia, removes this crucial regulatory brake, allowing neuroinflammation to proceed unchecked 3436.
The Dietary Inflammatory Index (DII)
The composition of the microbiome and the basal state of the immune system are heavily influenced by diet. Nutritional psychiatry utilizes the Dietary Inflammatory Index (DII) to assess the pro- and anti-inflammatory capacity of an individual's overall nutritional intake 3738. Diets with a high DII score - typically Western diets rich in ultra-processed foods (UPFs), saturated fats, and refined sugars - are associated with significant elevations in circulating CRP, IL-6, and TNF-α 3739.
Cross-sectional and dose-response meta-analyses demonstrate a linear relationship between elevated DII scores and the severity of psychiatric symptoms. Individuals in the highest quintile of dietary inflammation possess nearly double the risk of developing clinical depression, anxiety, and psychological distress compared to those in the lowest quintile 373840. Notably, individuals with severe mental illnesses consume significantly more pro-inflammatory foods and fewer anti-inflammatory nutrients, exacerbating their baseline inflammatory biotype 3241. Conversely, adherence to anti-inflammatory dietary patterns, such as the Mediterranean diet, has been shown in randomized controlled trials to significantly reduce depressive symptoms and peripheral inflammation 4142.
Table 2: Environmental Drivers of Systemic and Neural Inflammation
| Driver Category | Specific Factors | Biological Mechanism of Action | Psychiatric/Neurological Outcome |
|---|---|---|---|
| Dietary Intake | High DII Scores (Ultra-processed foods, low fiber). | Induces dysbiosis; starves SCFA-producing bacteria; promotes low-grade systemic inflammation. | Dose-response increase in incidence of depression, anxiety, and general distress 3842. |
| Microbiome | Reduction in Lactobacillus and Bifidobacterium. | Decreased butyrate production; compromised intestinal barrier (leaky gut); LPS translocation. | Heightened neuroinflammation, microglial activation, and impaired hippocampal neurogenesis 333436. |
| Latent Viruses | Cytomegalovirus (CMV), Epstein-Barr Virus (EBV). | Viral neurotropism; periodic reactivation via stress; chronic induction of anti-viral immunity. | Increased ratio of reactive microglia in prefrontal cortex; gray matter reduction; elevated suicide risk 434445. |
| Environmental Toxins | Air pollution, diesel fumes, wildfire smoke. | Induces oxidative stress and reactive astrogliosis; compromises BBB function. | Acute to chronic neuroinflammation; exacerbates PTSD, anxiety, and cognitive decline 46. |
Viral Pathogens and Post-Infectious Neuroinflammation
Pathogen exposure constitutes another significant environmental driver of neuroinflammation. Specific neurotropic viruses are capable of establishing lifelong latent infections that reactivate in response to physiological or psychological stress 4347. Cytomegalovirus (CMV) is particularly implicated in psychiatric pathology. Epidemiological data show that CMV seropositivity is significantly higher in populations with mood disorders and schizophrenia 4349. Postmortem analyses of CMV-positive individuals with MDD reveal severe neuroimaging abnormalities, including reduced gray matter volume, altered white matter integrity, and a substantially increased ratio of non-ramified (reactive) to ramified (resting) microglia within the dorsolateral prefrontal cortex 4345.
Similarly, Epstein-Barr Virus (EBV) and tick-borne pathogens have been identified as triggers for infection-induced autoimmune encephalopathies, wherein the immune system generates antibodies that cross-react with neuronal surface proteins 4448. The recent SARS-CoV-2 pandemic underscored the profound psychiatric impact of acute viral infections. The virus provokes a severe systemic cytokine storm that damages the cerebral endothelium and incites microglial reactivity. This results in lasting neurological sequelae, including cognitive impairment, anxiety, and treatment-resistant depression, demonstrating how acute pathogenic events can initiate chronic neuroinflammatory cascades 4749. It should be noted, however, that 12-month cohort studies tracking patients post-infection (e.g., comparing COVID-19 to Hepatitis C and Tick-Borne Encephalitis) indicate that psychiatric outcomes are more closely linked to the severity of the host's generalized inflammatory response rather than the specific type of virus contracted 4849.
Targeted Therapeutics and Precision Psychiatry Trials
The robust validation of the inflammatory biotype has accelerated the translation of neuroimmunological research into clinical practice. Over the past few years, numerous trials have explored the efficacy of repurposing immunomodulatory and anti-inflammatory agents to treat psychiatric and cognitive disorders. The critical finding across these trials is the absolute necessity of patient stratification; anti-inflammatory agents generally do not outperform placebos in unselected cohorts but demonstrate profound efficacy in biomarker-enriched subgroups.
Anti-Inflammatory Adjuncts in Depression
Comprehensive meta-analyses conducted in 2024 and 2025 reviewed randomized controlled trials utilizing pharmacological anti-inflammatory treatments for depression. When trials explicitly recruited depressed individuals utilizing an established cutoff for elevated inflammation (CRP ≥ 2 mg/L), anti-inflammatory treatments safely and effectively reduced depressive symptom severity (Hedges' g=0.35) and specifically targeted anhedonia (Hedges' g=0.40) 50. However, these agents did not significantly increase absolute remission rates compared to placebo, indicating that inflammation is a critical, but not singular, component of the disease architecture 50.
Two specific agents highlight this precision medicine approach: * Minocycline: A highly penetrant tetracycline antibiotic with distinct anti-inflammatory properties, minocycline operates centrally by inhibiting microglial activation and downregulating the production of IL-1β and TNF-α. In the randomized MINDEP trial, the addition of minocycline to standard antidepressant therapy resulted in significantly greater improvements in Hamilton Depression Rating Scale (HAM-D) scores, but this benefit was observed exclusively in patients presenting with elevated baseline CRP and IL-6 levels 51. * Infliximab: A chimeric monoclonal antibody that binds and neutralizes systemic TNF-α, preventing it from breaching the BBB. While infliximab failed to show efficacy in general populations with treatment-resistant depression, post-hoc analyses and subsequent targeted trials demonstrated significant antidepressant efficacy - particularly in resolving anhedonia and psychomotor retardation - in the specific subgroup of patients with highly elevated baseline inflammation (hs-CRP > 5 mg/L) 25253.
Modulating Neuroinflammation in Cognitive Decline
In the field of neurodegeneration, particularly Alzheimer's disease (AD), therapeutic strategies have increasingly recognized that clearing protein aggregates is insufficient without concurrent modulation of glial reactivity. In 2024 and 2025, the FDA approval of anti-amyloid monoclonal antibodies, such as lecanemab and donanemab, revolutionized early-stage AD treatment. Newer iterations, such as trontinemab, utilize "brain shuttle" technology to cross the BBB more effectively to clear amyloid plaques 5455. However, researchers note that managing the subsequent neuroinflammatory response remains a critical hurdle.
Preclinical trials are exploring novel combination therapies to address this. For example, a 2025 study paired low-dose Δ9-tetrahydrocannabinol (THC) with celecoxib, a selective COX-2 inhibitor, in a mouse model of Alzheimer's disease. While THC alone provided neuroprotective effects by reducing amyloid and tau pathology, it simultaneously increased certain inflammatory markers that counteracted cognitive benefits. However, when combined with the anti-inflammatory action of celecoxib, the combination effectively suppressed the neuroinflammatory signal, resulting in measurable improvements in memory and cognition 5657.
Additionally, agents traditionally utilized for psychiatric symptom control are being evaluated for their immunomodulatory side profiles. Lumateperone, an atypical antipsychotic, has demonstrated significant anti-inflammatory properties by directly modulating the NLRP3 inflammasome complex alongside its primary action on glutamatergic and dopaminergic receptors 58. These multifaceted pharmacological approaches highlight the future of neurotherapeutics: simultaneous targeting of neurotransmitter systems, protein aggregation, and the underlying neuro-immune cascade.
Conclusion
The extensive body of contemporary neuroimmunological research confirms that neuroinflammation is a fundamental driver of depression, anxiety, and cognitive decline. The traditional boundary between the peripheral immune system and the central nervous system is highly permeable, with systemic inflammation routinely breaching the brain via degraded endothelial tight junctions, active cytokine transport, and high-speed vagal nerve signaling. Once internal, these immune signals hyperactivate resident microglia and astrocytes, generating a neurotoxic environment that depletes vital neurotransmitters, halts synaptogenesis, and accelerates neuronal apoptosis.
Crucially, this pathology does not apply uniformly to all psychiatric patients. The validation of an "inflammatory biotype" - identifiable through peripheral biomarkers like CRP, IL-6, and specific leukocyte profiles - demands a shift toward precision psychiatry. Environmental factors such as microbiome dysbiosis, high-DII diets, and latent viral infections act as the primary kindling for this systemic inflammation, offering clear targets for lifestyle and medical intervention. As clinical trials increasingly demonstrate the efficacy of agents like minocycline and targeted monoclonal antibodies exclusively in biomarker-enriched subgroups, it is evident that the future of psychiatric and neurodegenerative care relies on simultaneously addressing the mind and the immune system.