Myelin Plasticity and White Matter Remodeling in Learning and Disease
Foundational Concepts in Central Nervous System Myelination
For over a century, the scientific consensus regarding central nervous system (CNS) myelin was heavily influenced by early histological mappings, such as those pioneered by Paul Flechsig in the early 20th century 1. Myelin, the lipid-rich, multilamellar membrane extended by oligodendrocytes, was conceptualized almost exclusively as a static and inert electrical insulator. Its primary established function was the facilitation of rapid, saltatory conduction of action potentials along axons, minimizing signal dissipation and reducing the metabolic cost of neuronal firing 2345. The traditional framework posited that myelination was a rigidly programmed developmental milestone. Once the CNS reached structural maturity in late adolescence or early adulthood, the myelin architecture was thought to remain largely fixed, undergoing subsequent macroscopic changes only under pathological conditions of demyelination or progressive neurodegeneration 234.
However, advanced neurobiological techniques - including in vivo longitudinal multiphoton imaging, sophisticated genetic fate-mapping, and high-resolution spatial proteomics - have catalyzed a profound paradigm shift 67. The white matter of the adult brain is now recognized as a highly dynamic and actively regulated tissue 34. Myelin plasticity, defined as the capacity of myelin-forming cells to continuously adapt their structure, volume, and topographical distribution in response to environmental stimuli, behavioral experience, and local neuronal activity, has emerged as a fundamental physiological mechanism of lifelong neuroplasticity 268.
Oligodendrocyte precursor cells (OPCs) - a population of multipotent, NG2-positive glial cells that comprise approximately 5% of all cells in the adult CNS - persist widely throughout the brain and spinal cord 59. These resident precursors retain the capacity to proliferate and differentiate into mature, myelinating oligodendrocytes across the entire human lifespan 49. This continuous oligodendrogenesis allows for the ongoing structural refinement of neural circuits long after primary development has ceased. Myelinated axons act not merely as passive biological cables but as highly calibrated conduction conduits. Subtle, targeted adjustments to myelin sheath thickness, the length of individual internodes, and the architecture of the unmyelinated nodes of Ranvier can fundamentally alter the timing, synchronization, and metabolic efficiency of complex neural networks 5810.
Spatial Scales and Axo-Myelin Topography
The functional impact of myelination is rigidly dictated by its microscopic spatial geometries. Within the CNS, myelin is not distributed uniformly. A fully mature oligodendrocyte can extend processes to ensheath up to 50 distinct axonal segments simultaneously, yet the specific dimensions of each sheath are highly variable and customized to the local biophysical environment 1112.
| Spatial Parameter | Typical Dimension / Threshold | Functional Implication in the CNS |
|---|---|---|
| Internode Length | 25 μm to 180 μm (Average ~101 μm in L5 pyramidal neurons) | Determines the distance an action potential travels passively before regeneration; longer internodes generally increase conduction velocity 1314. |
| Node of Ranvier Width | < 1.5 μm (typically 0.5 to 1.5 μm) | Highly concentrated zones of Nav1.6 and Kv1 channels; tight regulation of width is crucial for maintaining saltatory conduction integrity 131415. |
| Axon Diameter Threshold | > 0.3 μm (CNS minimum) | Oligodendrocytes generally do not myelinate fibers smaller than 0.3 μm due to strict mechanosensitive and biophysical constraints 1516. |
| g-ratio | ~0.60 to 0.75 (e.g., 0.698 for L5 main axon) | The ratio of inner axonal diameter to the total outer diameter of the myelinated fiber; optimizes the trade-off between conduction speed and spatial volume 414. |
| Analog-Digital Facilitation (ADF) Extent | 250 μm (unmyelinated) vs. up to 3,000 μm (myelinated) | Myelination increases the axonal length constant up to three-fold, expanding the spatial reach of subthreshold somatic membrane potential fluctuations to distant synapses 1314. |
The exact distribution of these sheaths is highly heterogeneous and axon-specific, particularly within the cerebral cortex. For example, among cortical GABAergic interneurons, fast-spiking parvalbumin-positive (PV) interneurons are heavily myelinated. Their myelination topography typically features short internodes clustered closely together, frequently punctuated by complex axonal branch points 1516. The myelination of these short-range axons is thought to facilitate and sustain rapid burst firing, which is critical for the function of cortical sensory inhibitory circuits 17. In sharp contrast, somatostatin-positive (SOM) interneurons, which possess thinner axonal shaft diameters (averaging ~0.303 μm) and shorter interbranch distances, are rarely myelinated 1516.
This precise topographical distribution is dictated in part by physical and mechanotransductive constraints. Oligodendrocytes rely on surface tension and mechanosensation to gauge the physical dimensions of target axons. Recent in vitro and in vivo studies have demonstrated that the mechanosensitive ion channel Piezo1 is actively localized in nascent myelin sheaths 18. Piezo1 detects the underlying fiber diameter and translates this mechanical tension into localized cellular signaling, instructing the elongation of the individual myelin segment to scale proportionately with the size of the enveloped axon 18.
Cellular and Molecular Mechanisms of Myelin Plasticity
The process by which the CNS adjusts its myelin architecture is highly intricate, relying on an active, continuous dialogue between firing neurons and the encompassing oligodendrocyte lineage. This cellular crosstalk translates transient electrical activity and chemical neurotransmission into enduring structural modifications in the white matter 219.
De Novo Myelination Versus Structural Remodeling
Current research indicates that myelin plasticity manifests through two distinct, yet complementary, cellular mechanisms: the de novo generation of new myelin sheaths on previously unmyelinated axonal segments, and the geometric structural remodeling of pre-existing, mature myelin sheaths 3410. The relative contribution of each mechanism to overall adult neuroplasticity remains a subject of ongoing debate within the field.
De novo myelination is primarily driven by the activation, targeted migration, proliferation, and terminal differentiation of OPCs into mature oligodendrocytes. During the acquisition of a new motor skill or in response to artificially heightened neuronal activity, local populations of OPCs are recruited to active neural circuits. These precursors engage naked axons, differentiate, and generate entirely new internodes 292021. This sequence requires overcoming endogenous inhibitory signals in the local microenvironment, establishing secure axo-glial contact, and dramatically expanding the oligodendrocyte plasma membrane to form multiple concentric wrappings 1021.
Conversely, emerging evidence suggests that the physical remodeling of pre-existing myelin constitutes a major, if not dominant, form of adult plasticity. Mature oligodendrocytes - once thought to be post-mitotic and structurally fixed - retain the lifelong capacity to alter the length and thickness of their established internodes in response to ongoing local neuronal firing 3410. For instance, physiological studies utilizing environmental enrichment paradigms (which naturally stimulate extensive neuronal activity) have demonstrated that the corpus callosum and somatosensory cortex in young adult mice exhibit substantial enlargement of callosal axon diameters accompanied by a corresponding increase in the thickness of pre-existing myelin sheaths 22. Notably, this activity-dependent thickening occurred without a statistically significant upregulation in OPC proliferation or the de novo generation of new myelin on unmyelinated segments 22. These findings imply that in the fully developed adult CNS, biophysical or energetic constraints may limit maximal de novo production, elevating the progressive thinning or thickening of originally existing mature (OEM) myelin to a primary mode of adaptive plasticity 422.
Transcriptional Control and Epigenetic Regulation
Beneath these macroscopic cellular changes lies a highly coordinated layer of transcriptional and epigenetic regulation. The transition of an OPC to a mature oligodendrocyte is governed by an orchestrated sequence of transcription factors, most notably the SOX family (SOX10) and OLIG factors (OLIG1, OLIG2) 2324. In adulthood, these regulatory networks continue to dictate myelin maintenance.
Recent research has identified specific epigenetic repressor systems that naturally slow or halt myelin production to maintain homeostasis. For example, the protein Tfii-i (encoded by the Gtf2i gene) functions as a natural brake in the brain, binding to regulatory elements to limit the activity of Sox10 and Mbp 24. Experimental deletion of Tfii-i in mouse models results in the generation of abnormally thick myelin sheaths and significantly enhanced electrical conduction speeds, revealing a baseline level of continuous epigenetic suppression that actively prevents hyper-myelination 24. Similarly, DNA methylation and histone modifications (such as H3K27me3) act at state-specific enhancers to govern transcriptomic state switches in both the oligodendrocyte and microglial lineages across development, aging, and disease contexts 725.
Activity-Dependent Regulation and Signaling Pathways
The spatial targeting of myelination is not arbitrary; it is exquisitely tuned to neuronal activity. Neurons that exhibit higher firing rates are preferentially myelinated, ensuring that frequently utilized circuits are optimized for speed, precision, and metabolic efficiency 2102025.
Evidence from Optogenetic and Chemogenetic Models
Experimental paradigms utilizing optogenetics and Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) have provided conclusive evidence for activity-dependent axo-glial coupling. In vivo optogenetic stimulation of cortical layer V projection neurons - typically via the expression of the excitatory opsin Channelrhodopsin-2 (ChR2) - directly induces localized increases in OPC proliferation, oligodendrogenesis, and subsequent myelin thickness along the specific stimulated tracts 4112126.
Chemogenetic methodologies have yielded corroborating results. Prolonged chemogenetic activation of retinal ganglion cells via the Gq (hM3Dq) pathway utilizing clozapine (the active metabolite of CNO in vivo) results in a measurably decreased average g-ratio within the optic nerve, indicating the deposition of thicker myelin specifically around the active axons 419. Furthermore, these structural adaptations appear to be bidirectional; suppressing neuronal activity via the inhibitory Gi (hM3Di) DREADD pathway leads to observable deficits or thinning in myelin maintenance 19.
Glutamatergic and Neurotrophic Signaling
To translate transient electrical action potentials into long-term cellular remodeling, oligodendroglia rely on a complex array of chemical intermediaries. Glutamatergic neurotransmission serves as a primary conduit for this neuron-oligodendrocyte communication. Both OPCs and mature oligodendrocytes express functional AMPA, NMDA, and metabotropic glutamate receptors, through which they monitor local synaptic and axonal firing 1021.
Recent high-resolution, time-lapse imaging utilizing zebrafish models has definitively identified metabotropic glutamate receptor 5 (mGluR5) as a critical molecular mediator of activity-driven myelin sheath growth 27. Pharmacological activation of mGluR5 promotes the rapid elongation of myelin sheaths during development, whereas specific genetic or pharmacological inhibition of this receptor restricts sheath growth without broadly impacting the survival or proliferation of the broader oligodendrocyte lineage 27. This indicates a direct, receptor-mediated mechanism by which excitatory neurotransmission physically dictates the expansion of myelin coverage.
Beyond neurotransmitters, neurotrophic factors and complex intracellular signaling cascades regulate the myelinating capacity of glial cells. Brain-derived neurotrophic factor (BDNF), which is secreted from neurons in an activity-dependent manner, binds to TrkB receptors situated on oligodendrocyte lineage cells 4212829. This binding event promotes OPC maturation, enhances cell survival, and upregulates the transcription of myelin structural proteins. Intracellularly, these extracellular signals converge on the Akt-mTOR (mammalian target of rapamycin) signaling pathway, a central metabolic hub that governs myelin protein translation and lipid synthesis.
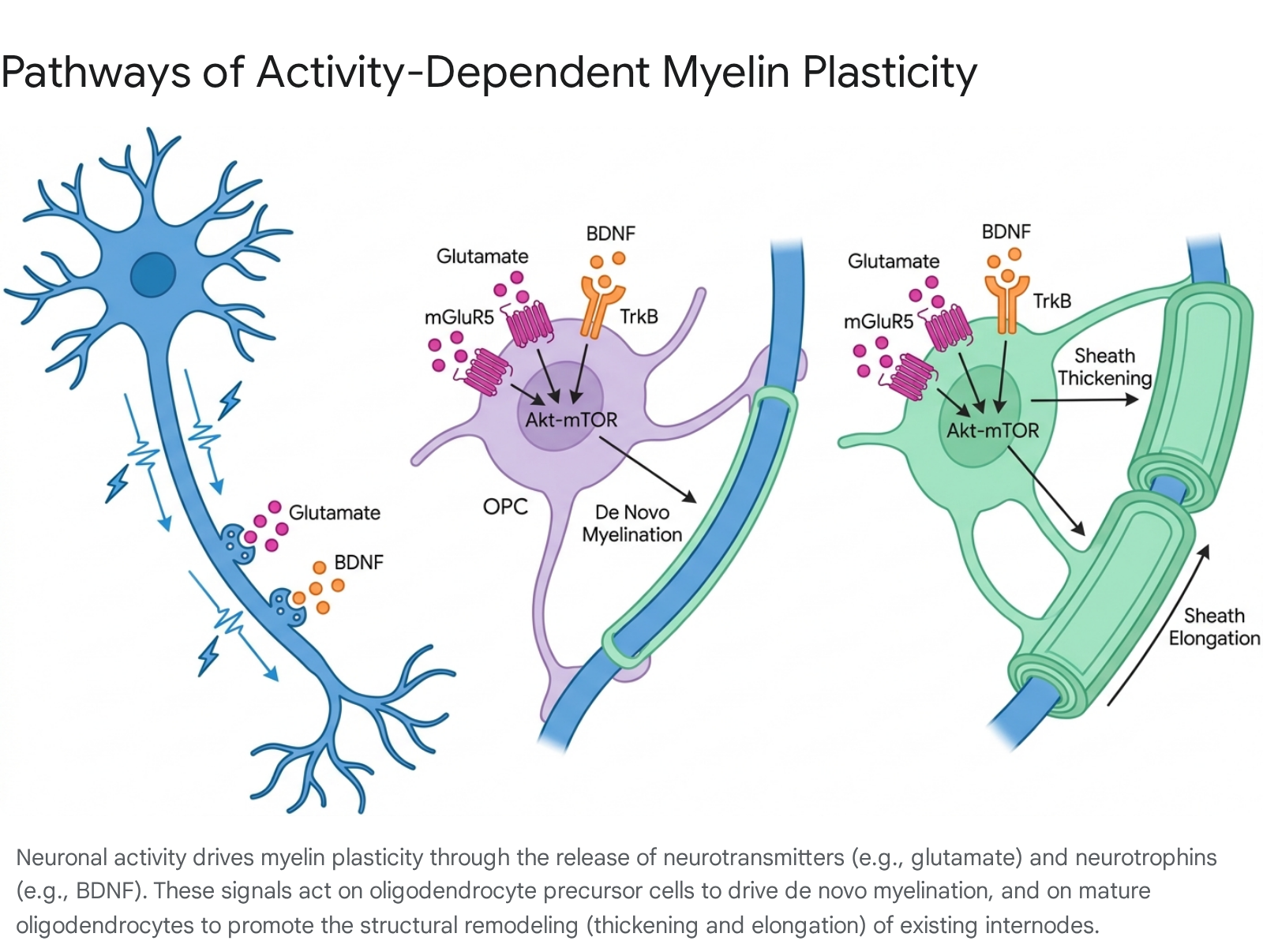
The critical nature of this hub is illustrated by experiments involving 3-phosphoinositide-dependent protein kinase-1 (PDK1), an upstream regulator of Akt-mTOR. Conditional, targeted ablation of PDK1 in OPCs results in profound deficits in postnatal hippocampal myelination, stalled oligodendrogenesis, and severe impairments in learning and memory 30.
Glial Crosstalk and the Microenvironment
Myelin plasticity does not occur in a cellular vacuum. It is heavily modulated by the broader glial microenvironment, which includes dynamic populations of astrocytes and microglia that continuously assess and react to metabolic and inflammatory cues 249.
Microglial States in White Matter Homeostasis
Microglia, the resident immune cells of the CNS, possess diverse, context-dependent transcriptomic states that exert significant influence over white matter integrity. Single-cell RNA sequencing and genetic fate-mapping have identified distinct microglial subpopulations, including proliferative-region-associated microglia (PAM), which are enriched in developing white matter tracts like the corpus callosum 725. Throughout the lifespan, these microglial populations exhibit remarkable transcriptomic and epigenomic plasticity, transitioning into disease-associated microglia (DAM) during neurodegenerative states, or white matter-associated microglia (WAM) during normal aging 725.
These distinct microglial states heavily influence the myelinating capacity of adjacent oligodendrocytes. M2-polarized (alternatively activated) macrophages and appropriately regulated microglia secrete anti-inflammatory cytokines (such as IL-10) and neurotrophic factors (like IGF-1) that actively promote OPC differentiation and support myelin sheath formation 2831. Conversely, chronic neuroinflammation, characterized by M1-polarized (classically activated) macrophages and the release of pro-inflammatory cytokines like IFN-γ and TNF-α, actively suppresses OPC differentiation, promotes premature OPC apoptosis, and accelerates the degradation of the existing myelin architecture 283132.
Metabolic Influences and Lipid Dysregulation
The glial microenvironment is highly sensitive to systemic metabolic health. For instance, the consumption of high-fat Western diets induces peripheral hyperlipidemia, which significantly alters microglial functionality and exacerbates myelin vulnerability 33. Lipid dysregulation, often mediated by variations in apolipoprotein E (ApoE) transport, impairs the capacity of microglia to maintain local white matter homeostasis 33. In murine models of Alzheimer's disease, diet-induced hypercholesterolemia drives microglial dysfunction, weakening their ability to clear amyloid-beta plaques and actively exacerbating neuroinflammation and collateral myelin degradation 33. Thus, the maintenance of systemic metabolic health appears inextricably linked to the preservation of adaptive myelin plasticity in the aging CNS.
Integration of Myelin Plasticity in Cognitive and Motor Function
While traditional neuroscience focused primarily on synaptic potentiation as the sole physical substrate for learning and memory, it is now firmly established that synaptic plasticity acts in tight concert with myelin plasticity to coordinate complex, long-term behavioral adaptations 610.
Motor Skill Acquisition and Circuit Maturation
Motor learning provides one of the most robust and highly studied paradigms for observing adaptive myelination. The acquisition of complex, novel motor skills - such as a mouse learning to navigate an irregular, complex running wheel, or a human mastering the piano - requires the precise, millisecond-level temporal integration of extensive sensory and motor circuits 11202534.
Following the initiation of motor training, there is a rapid upregulation of OPC proliferation and a subsequent surge in terminal differentiation into mature oligodendrocytes within highly engaged white matter tracts, including the primary motor cortex and the corpus callosum 1126. The absolute necessity of this process for learning has been definitively proven using genetic ablation strategies. By blocking the differentiation of new oligodendrocytes (e.g., via the conditional deletion of the transcription factor myrf or the targeted inhibition of Nf1 exclusively in OPCs), researchers can fundamentally impair the acquisition and long-term consolidation of these complex motor tasks, even if synaptic plasticity remains largely intact 2026. Interestingly, this plasticity is temporally biphasic; during the initial exploratory phases of learning, pre-existing myelin sheaths may transiently retract to create an intermittent myelination pattern. This structural loosening is subsequently filled by the de novo addition of new sheaths covering large unmyelinated gaps once the skill becomes permanently consolidated 34.
Memory Consolidation and Network Synchronization
Beyond basic motor output, myelin remodeling is vital for higher-order cognitive processing, spatial memory encoding, and sleep-dependent memory consolidation 23435. Efficient cognitive function relies intrinsically on the precise synchronization of distant neuronal ensembles, a biophysical feature highly dependent on the conduction delays strictly modulated by myelin thickness and internodal spacing 1034.
Advanced computational models of spiking neurons demonstrate that simple, biologically grounded rules for myelin learning - where highly active, reverberating neural pathways experience a localized decrease in conduction delays due to targeted myelin thickening - can independently enable neural circuits to learn associations without requiring any concurrent modifications to synaptic weights 20. Physiologically, this velocity tuning allows for the phase-amplitude coupling of distinct brain rhythms. For example, successful contextual fear conditioning relies heavily on the precise temporal coupling between spindle oscillations generated in the prefrontal cortex and sharp wave ripples propagating from the hippocampus, an interaction dependent on optimal white matter integrity 3437.
Recent human studies leveraging simultaneous polysomnography and ultrahigh-field magnetic resonance spectroscopy suggest that myelin plasticity and memory transfer are intimately tied to sleep architecture. Memory traces appear to undergo rapid shifts in circuit stability during sleep: memories enter a highly malleable, transiently unstable state during non-rapid eye movement (NREM) sleep (facilitated by slow wave-spindle coupling) before being hyper-stabilized and transferred into permanent long-term storage during phasic REM sleep 35. This intricate knowledge integration process is hypothesized to be underwritten by rapid, state-dependent glial and synaptic remodeling.
Pathological Consequences of Maladaptive Myelination
If optimal brain function requires tightly regulated, adaptive myelination, it follows that the dysregulation of these exact processes leads to profound neurological, cognitive, and psychiatric consequences. Myelin plasticity can "go wrong" through a failure to myelinate appropriately during critical developmental windows, progressive structural degradation during aging, or the maladaptive hyper-myelination of destructive, pathological circuits 52336.
Schizophrenia and Bipolar Disorder
Psychotic spectrum disorders (PSD), principally encompassing schizophrenia and bipolar disorder, have increasingly been reframed by neurobiologists as diseases of macroscale brain "disconnectivity." This disconnectivity is fundamentally driven by subtle, widespread white matter and oligodendroglial dysfunction 233738.
Extensive postmortem analyses of both schizophrenic and bipolar brains consistently reveal significant downregulation across the primary structural proteins of the axo-myelin unit, including reductions in proteolipid protein (PLP), myelin basic protein (MBP), 2',3'-Cyclic-nucleotide 3'-phosphodiesterase (CNP), and myelin oligodendrocyte glycoprotein (MOG) 2336. This structural deficiency is mirrored at the transcriptomic level by severe reductions in the expression of critical oligodendroglial lineage transcription factors, such as OLIG1, OLIG2, and SOX10 2336. Furthermore, dysregulation of genes associated with oligodendrocyte maturation and synaptic plasticity, such as p57Kip2, NRG1, and specific single-nucleotide polymorphisms (SNPs) in the DDR1 gene, represent significant genetic risk factors for the development of schizophrenia 233638. Advanced multimodal MRI imaging further corroborates these cellular deficits, firmly linking specific microstructural alterations in myelin-axonal geometry to diminished working memory capacity and the severity of positive symptoms in early-stage PSD 3738.
Mechanistically, disruptions in the splicing of specific proteins like hnRNP A1 have been shown to directly interfere with myelin stability and the overall brain remyelination proteome, presenting novel molecular targets that link genetic vulnerability to observable demyelinating phenotypes in psychiatric disease 39.
Autism Spectrum Disorder
Similar hypomyelination phenotypes are increasingly observed in Autism Spectrum Disorder (ASD). The interplay between myelin plasticity and sensory processing deficits is clearly illustrated in the NL3-R451C-KI mouse model of ASD 1740. In these subjects, severe localized deficits in OPC differentiation and mature oligodendrocyte generation are restricted to specific regions, notably the barrel cortex 1740.
This targeted hypomyelination significantly impairs the electrical excitability of local parvalbumin (PV) interneurons, leading to a disruption in local gamma neuronal oscillations 1740. Functionally, this micro-architectural defect fundamentally alters sensory discrimination, resulting in impaired texture recognition behaviors 1740. Similarly, genetic models such as the Smg5-CKO mouse demonstrate that disruptions in nonsense-mediated mRNA decay pathways dramatically attenuate the expression of essential myelin genes (Mbp, Plp1, Mag), further establishing the critical link between genetic neurodevelopmental mutations, disrupted myelin plasticity, and pervasive functional impairments 41.
Addiction, Opioid Reward, and Maladaptive Reinforcement
Neuroplasticity is inherently value-neutral; the mammalian brain will structurally adapt to reinforce whatever neural circuits are highly active, even if the resulting behavior is biologically destructive. Groundbreaking recent research highlights that adaptive myelination plays a central, insidious role in the pathology and persistence of drug addiction 25.
Administration of exogenous opioids (such as morphine) or psychostimulants (like cocaine), or even the direct optogenetic stimulation of endogenous dopamine-producing neurons, selectively triggers a rapid, massive proliferation of OPCs specifically within the ventral tegmental area (VTA) - the anatomical core of the brain's reward circuitry 2544. This drug-induced neuronal hyper-activity drives localized myelin plasticity, physically tuning the circuit to become hyper-responsive to the substance 44.
When researchers genetically engineered mice to block this de novo oligodendrogenesis specifically within the VTA, the subjects exhibited dampened dopamine release dynamics in the target nucleus accumbens 44. Crucially, these mice were unable to learn or remember the location of their morphine reward, failing to develop standard addictive behavioral conditioning 2544. This paradigm-shifting discovery suggests that the enduring, intractable nature of substance-use disorders is partially embedded within the physical white matter structure of the reward pathway. Consequently, targeting myelin plasticity could offer unprecedented novel interventions to physically "unlearn" addiction by promoting the bidirectional down-regulation of maladaptive myelin 25. Similarly, maladaptive myelination has been shown to exacerbate epileptic seizure disorders; the extreme neuronal firing during an initial seizure drives pathological myelin remodeling that subsequently facilitates faster, more synchronized, and significantly more severe future seizures 25.
Aging and Cognitive Decline
Even in the absence of acute disease, the endogenous capacity for myelin plasticity wanes steadily with age, contributing heavily to normative cognitive decline and enhancing vulnerability to subsequent neurodegenerative diseases 58. In the aging brain, previously stable mature myelin sheaths often undergo spontaneous decompaction, blebbing, and fragmentation 5.
Over decades, the accumulation of this myelin debris overwhelms the phagocytic clearance capacity of local microglia and macrophages. This leads to the establishment of a chronically inhibitory microenvironment that not only impairs the de novo differentiation of new OPCs but actively prevents the necessary length-remodeling and maintenance of existing sheaths 5. Therefore, age-related cognitive slowing is likely driven, in part, by the gradual loss of precision in analog-digital signal facilitation due to degrading white matter integrity 58.
Demyelination, Remyelination, and Axonal Regeneration
While normative aging involves a slow, insidious decline in myelin plasticity, frank demyelinating diseases like Multiple Sclerosis (MS) are characterized by the catastrophic, acute, and immune-mediated destruction of the myelin sheath.
Demyelination Dynamics in Multiple Sclerosis
In MS, autoreactive peripheral immune cells infiltrate the CNS and erroneously strip myelin from structurally intact axons. This loss of insulation drastically increases membrane capacitance, leading to slowed, dispersed, or completely blocked electrical signal transmission 24324243. Clinically, this presents as acute episodes of sensory loss, motor weakness, visual impairment, and profound cognitive fatigue 2444.
Initially, the CNS mounts a vigorous, spontaneous remyelination response. OPCs are rapidly recruited to the site of the inflammatory lesion (the "plaque") 93242. However, as the disease progresses from its relapsing-remitting phase into the chronic or secondary progressive phases, this endogenous repair mechanism systematically fails 932.
Crucially, the failure of remyelination in chronic MS is rarely due to an absolute depletion or absence of OPCs. Instead, histological analyses reveal that OPCs successfully migrate to the lesion site but remain stalled indefinitely in a quiescent, pre-myelinating state, fundamentally unable to undergo terminal differentiation 32424546. This developmental arrest is driven by a highly toxic, inhibitory pathological microenvironment laden with un-cleared myelin debris, inflammatory cytokines, and specific inhibitory molecular signals - such as Semaphorin 3A and LINGO-1 - that actively suppress the complex transcriptional programs required for myelin generation 92842. Left permanently uninsulated, these chronically demyelinated axons lose vital glial metabolic support, leaving them highly vulnerable to progressive, irreversible axonal degeneration. It is this secondary axonal death, rather than the initial demyelination, that is the primary driver of permanent, accumulating clinical disability in progressive MS 93247.
Distinguishing Remyelination from Axonal Regeneration
In therapeutic and clinical contexts, it is absolutely critical to distinguish the biological process of remyelination from axonal regeneration, though both ultimately aim to restore functional neural circuitry 93148.
Axonal regeneration involves the actual biological regrowth and physical extension of a severed neuronal projection (e.g., following a traumatic spinal cord injury) across a highly inhibitory glial scar environment to re-establish a synaptic connection with a distant target 93148. Remyelination, conversely, involves only the glial re-insulation of an axon that remains structurally intact but has been pathologically denuded of its myelin 932.
While modern high-efficacy MS disease-modifying therapies (DMTs) like ocrelizumab and fingolimod efficiently suppress the peripheral immune system, prevent relapses, and halt the formation of new inflammatory lesions, they exert almost zero impact on either remyelination or axonal regeneration within existing, established chronic lesions 4349. Consequently, discovering mechanisms to overcome the inhibitory microenvironment and directly stimulate stalled OPCs represents the most significant unmet medical need in clinical neurology today 4447.
The Therapeutic Pipeline for Remyelination
The urgent race to develop drugs that directly stimulate OPC differentiation and facilitate profound myelin repair has yielded a diverse, highly innovative pipeline of small molecules, repurposed biologics, and targeted pathway inhibitors currently undergoing rigorous preclinical and clinical evaluation.
Clinical Pipeline of Remyelinating Agents
The data table below summarizes the most prominent and promising agents currently under active investigation for their remyelinating potential in multiple sclerosis and related demyelinating conditions, tracking their progression from preclinical validation to human trials.
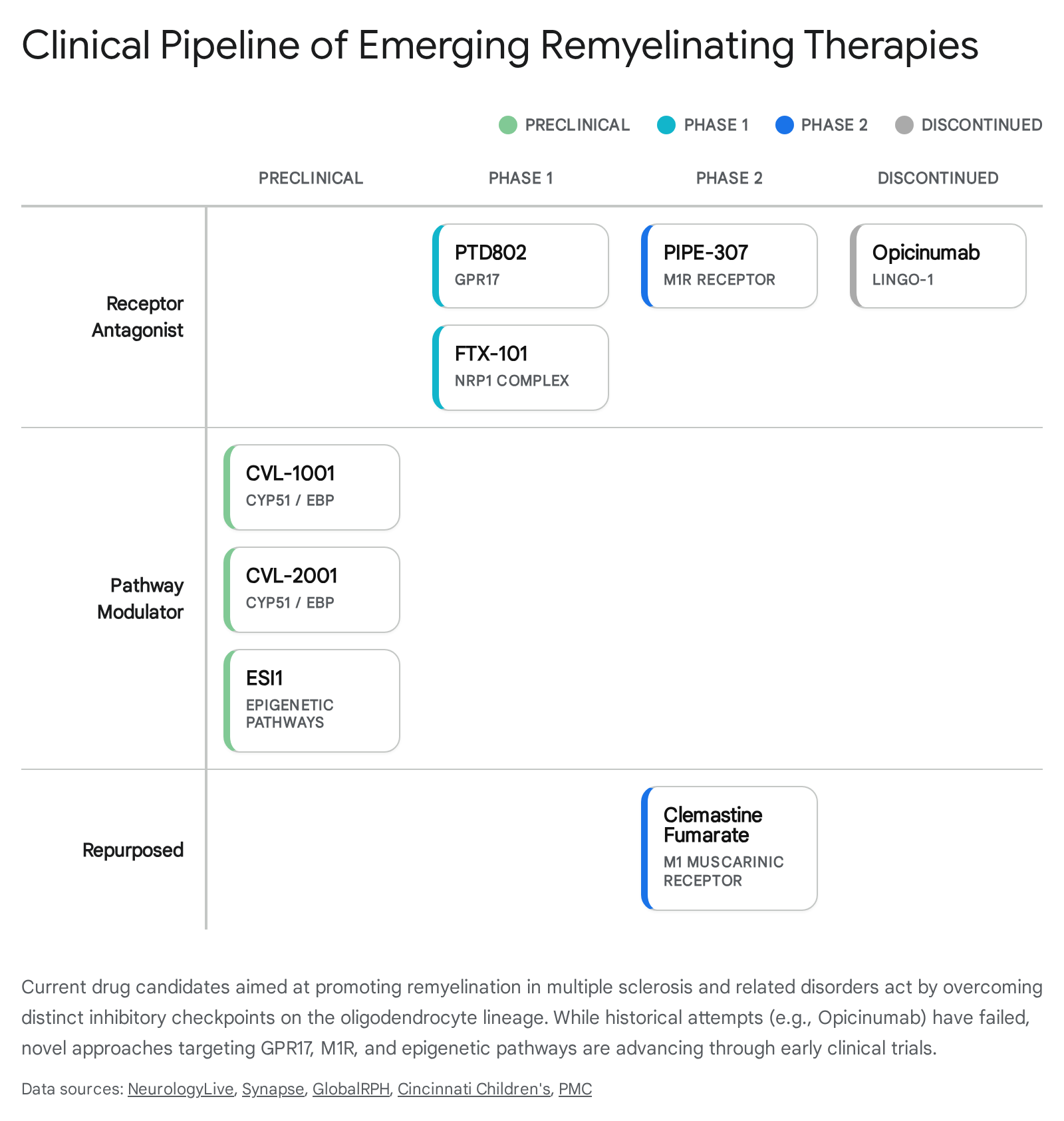
| Drug Candidate | Sponsor / Developer | Mechanism of Action (Target) | Current Clinical Phase | Key Details / Outcomes |
|---|---|---|---|---|
| PTD802 | Pheno Therapeutics | GPR17 Antagonist | Phase 1 (2025) | Releases the GPR17 "brake" on OPCs, allowing them to differentiate into mature oligodendrocytes. Cleared by UK MHRA for first-in-human healthy volunteer trials 424550515556. |
| PIPE-307 | Contineum Therapeutics / UCSF | Muscarinic M1 Receptor (M1R) Antagonist | Phase 2 | Blocks M1R on OPCs to promote differentiation. Demonstrated safety in Phase 1; preclinical models showed reversal of myelin loss 4244. |
| FTX-101 (Tasronetide) | Find Therapeutics | PlexinA1 / Neuropilin 1 (NRP1) Modulator | Phase 1 (2024-2025) | First-in-class synthetic peptide. Blocks inhibitory Semaphorin 3A signaling, disinhibiting OPC migration and differentiation. Targeted initially for chronic optic neuropathy 4252535954. |
| Clemastine Fumarate | Academic / Various | Antihistamine (M1 Muscarinic Antagonist effect) | Phase 2 (Completed/Ongoing) | Met primary endpoint in ReBUILD (optic neuropathy) and CCMR-Two (with metformin), significantly reducing VEP latency. Failed in TRAP-MS due to disability worsening 475556. |
| CVL-1001 / CVL-2001 | Convelo Therapeutics | CYP51 / EBP Inhibitor (Cholesterol Biosynthesis) | Preclinical / IND-enabling | Induces accumulation of specific sterol intermediates that powerfully drive OPC maturation, showing unique efficacy in aged/inflamed CNS models 4257. |
| ESI1 | Cincinnati Children's / UC | Epigenetic Silencing Inhibitor | Preclinical | Un-silences mature oligodendrocytes by increasing active H3K27ac marks and decreasing repressive H3K27me3/H3K9me3 marks. Shown to rapidly reverse age-related myelin loss in mice 4446585960. |
| Opicinumab | Biogen | Anti-LINGO-1 Monoclonal Antibody | Discontinued (2020) | Targeted LINGO-1 (an endogenous inhibitor of myelination). Failed to meet primary efficacy endpoints in large Phase 2 SYNERGY/RENEW trials, halting development 42. |
Receptor Antagonists and Pathway Modulators
The current vanguard of modern remyelination therapy focuses heavily on highly selective small molecules designed to block the specific receptors that prevent OPC differentiation in the lesion environment.
One of the most highly anticipated candidates is PTD802, developed by Pheno Therapeutics. PTD802 is an orally bioavailable, highly selective small-molecule antagonist of the G-protein coupled receptor 17 (GPR17). GPR17 acts as a physiological "molecular timer" or intrinsic brake on OPCs; it temporarily promotes their initial proliferation but must be firmly downregulated for the cell to terminally differentiate 4245. In chronic MS lesions, GPR17 expression remains aberrantly high, permanently stalling repair 45. By specifically antagonizing this receptor, PTD802 effectively releases the brake, driving stalled OPCs to mature and deposit new myelin 4555. In January 2025, PTD802 received official clearance from the UK MHRA to commence first-in-human Phase 1 clinical trials, marking it as the first selective GPR17 antagonist to enter human testing 505556.
Operating on a separate molecular axis, FTX-101 (tasronetide), developed by Find Therapeutics, represents another distinct approach. FTX-101 is a first-in-class synthetic peptide designed to modulate the PlexinA1/Neuropilin 1 (NRP1) transmembrane receptor complex 425253. This receptor complex mediates the destructive effects of Semaphorin 3A, a guidance cue that accumulates heavily in demyelinated lesions and actively repels OPC migration while halting differentiation 4252. By interfering with the heterodimerization of the PlexinA1/NRP1 complex, FTX-101 disinhibits the microenvironment, allowing OPCs to successfully migrate into the lesion core and differentiate 52. The drug entered Phase 1 single-ascending and multiple-ascending dose studies in healthy male volunteers between late 2024 and 2025, with an initial clinical focus set on treating chronic optic neuropathy 425259.
Other advanced approaches target intracellular lipid metabolism rather than surface receptors. CVL-1001 and CVL-2001 act by inhibiting specific cholesterol biosynthesis enzymes, namely sterol 14-demethylase (CYP51) and emopamil binding protein (EBP) 4257. The targeted inhibition of these specific enzymes causes an intracellular accumulation of unique sterol intermediates that powerfully stimulate the terminal differentiation of OPCs. This metabolic approach shows particular efficacy in overcoming the repair deficits typically associated with highly inflamed or aged CNS models, bypassing receptor-mediated blockades entirely 4257.
Overcoming Epigenetic Silencing in Mature Oligodendrocytes
While the majority of pipeline agents focus on stimulating precursor cells (OPCs), groundbreaking recent research highlights the profound therapeutic potential of reprogramming already-mature oligodendrocytes that reside near lesions. Epigenetic mapping studies conducted on postmortem MS tissues revealed a stunning discrepancy: mature oligodendrocytes within lesions uniformly lack an activating histone mark (H3K27ac) while massively overexpressing repressive epigenetic marks (H3K27me3 and H3K9me3) 585960. This signature effectively permanently silences their intrinsic repair and lipid-production genes.
In 2024, researchers at Cincinnati Children's Hospital identified a powerful small-molecule compound, ESI1 (epigenetic-silencing-inhibitor-1), capable of cleanly reversing this exact pathological profile 5859. In laboratory models, ESI1 exposure tripled the levels of activating H3K27ac while simultaneously slashing the repressive marks 5859. This epigenetic shift induced the formation of "biomolecular condensates" inside the cell nucleus - specialized hubs that supercharged the lipid and cholesterol production absolutely required for rapid myelin generation 465859. When tested in vivo in aged mice, MS mouse models, and highly complex human brain organoids, ESI1 treatment robustly regenerated thick myelin sheaths and demonstrably improved neurological function (such as water maze navigation) 444658. This breakthrough proves the concept that mature oligodendrocytes - not just undifferentiated precursor cells - can be pharmacologically un-silenced to drive massive remyelination, opening an entirely new class of epigenetic therapeutics 4446.
Clinical Endpoints and Biomarkers in Remyelination Trials
A significant historical hurdle in remyelination research has been the difficulty of conclusively demonstrating clinical efficacy in human trials. Biogen's Opicinumab (anti-LINGO-1) possessed a highly compelling biological rationale but failed to meet primary efficacy endpoints in its large Phase 2 trials (SYNERGY and RENEW) and was discontinued in 2020 42. This failure highlighted the extreme difficulty of measuring localized myelin repair in human patients against the noisy, fluctuating backdrop of ongoing MS inflammation and standard clinical disability scales.
To overcome this measurement barrier, modern clinical trials heavily rely on highly quantitative, objective electrophysiological biomarkers. Visual Evoked Potentials (VEP) latency has emerged as the gold-standard functional readout for early-phase remyelination studies. Because the human optic nerve is an extensively myelinated tract, demyelination (such as following optic neuritis) significantly delays the P100 electrical signal - a voltage peak that normally arrives at the occipital cortex exactly 100 milliseconds after a visual checkerboard stimulus 5561. Successful pharmacological remyelination predictably and measurably reduces this signal delay 4761.
This precise metric was crucial in validating the remyelinating potential of Clemastine fumarate, a repurposed, over-the-counter first-generation antihistamine with known off-target muscarinic (M1R) antagonist properties. In the landmark Phase 2 ReBUILD trial, clemastine significantly improved VEP latency by 1.7 milliseconds per eye compared to placebo in patients suffering from chronic demyelinating optic neuropathy 47. Most recently, in late 2025, the Phase 2 CCMR-Two trial (which tested clemastine combined with the diabetes drug metformin) reported a statistically significant 1.2-millisecond reduction in VEP latency over a six-month period, further solidifying the proof-of-concept that pharmacological remyelination is achievable and measurable in living humans 475556.
However, repurposed drugs like clemastine often carry severe, dose-limiting side effects (e.g., profound sedation) or present complex interactions depending on the specific disease stage. The TRAP-MS trial, for instance, actually had to halt its clemastine treatment arm after investigators observed unexpectedly rapid worsening of disability in non-lesional MS patients, underscoring the potential neurotoxic risks of off-target effects 5556. This highlights the absolute necessity for the highly selective, next-generation targeted agents (like PTD802 and PIPE-307) currently advancing through Phase 1 and 2 pipelines.