Mutation accumulation theory of aging
Evolutionary Gerontology Foundations
The existence of biological aging - formally defined as senescence - has historically presented a profound paradox for evolutionary biology. Natural selection acts to maximize the inclusive fitness of organisms, constantly optimizing phenotypic traits that enhance survival and reproductive success. It would logically follow that natural selection should favor physiological mechanisms capable of maintaining organismal integrity indefinitely, thereby maximizing the reproductive window of the organism. However, the vast majority of multicellular organisms undergo a progressive, inexorable decline in physiological function, reproductive capacity, and survival probability as chronological age advances 12.
The resolution to this evolutionary paradox began to emerge in the mid-twentieth century, shifting the scientific consensus away from the concept of "programmed death." Early theorists, such as August Weismann in the late nineteenth century, had erroneously posited that aging evolved for the "good of the species" to clear out older individuals and make room for younger generations 23. This group-selectionist argument is mathematically unstable in standard population genetics; any mutant individual that avoids aging would continue to reproduce, rapidly outcompeting its senescing peers and driving the "programmed death" allele to extinction 3. The modern evolutionary synthesis instead required a framework based strictly on individual selection.
The conceptual breakthrough originated from population geneticists J.B.S. Haldane and R.A. Fisher in the 1930s, who recognized that the force of natural selection is not uniform across an organism's lifespan. Haldane famously noted that a dominant genetic mutation causing a fatal disease like Huntington's chorea could remain highly prevalent in human populations precisely because its effects typically do not manifest until after the age of thirty 234. By that age, early humans had already achieved the bulk of their reproductive output. Therefore, the allele causing the disease is passed to the next generation before natural selection can eliminate the carrier. Building explicitly upon this demographic reality, British biologist Peter Medawar formally articulated the Mutation Accumulation (MA) theory of aging in 1952 25. Medawar proposed that aging is not an adaptive trait selected for its utility, but rather an inescapable byproduct of the declining force of natural selection with chronological age 25.
The Concept of the Selection Shadow
To comprehend Medawar's theory, one must isolate the concept of extrinsic mortality. In wild populations, virtually all organisms are subject to environmental hazards such as predation, starvation, infectious disease, and extreme weather. Because of these constant environmental pressures, the probability of an individual surviving to an advanced age is statistically low, regardless of the organism's intrinsic physiological robustness 67.
Because wild cohorts are continually decimated by extrinsic factors over time, the breeding population is predominantly composed of young individuals. Consequently, the later stages of life contribute minimally to the gene pool of the subsequent generation 467. If a genetic mutation arises that is unconditionally lethal in early development, natural selection will purge it from the population immediately, as the carrier will leave no offspring. However, if a mutation is entirely neutral during youth and reproductive maturity, but exerts severely deleterious effects only at an advanced age (e.g., beyond the typical life expectancy of the species in the wild), natural selection is virtually powerless to act against it.
The mutation effectively resides in the "selection shadow" 4789.
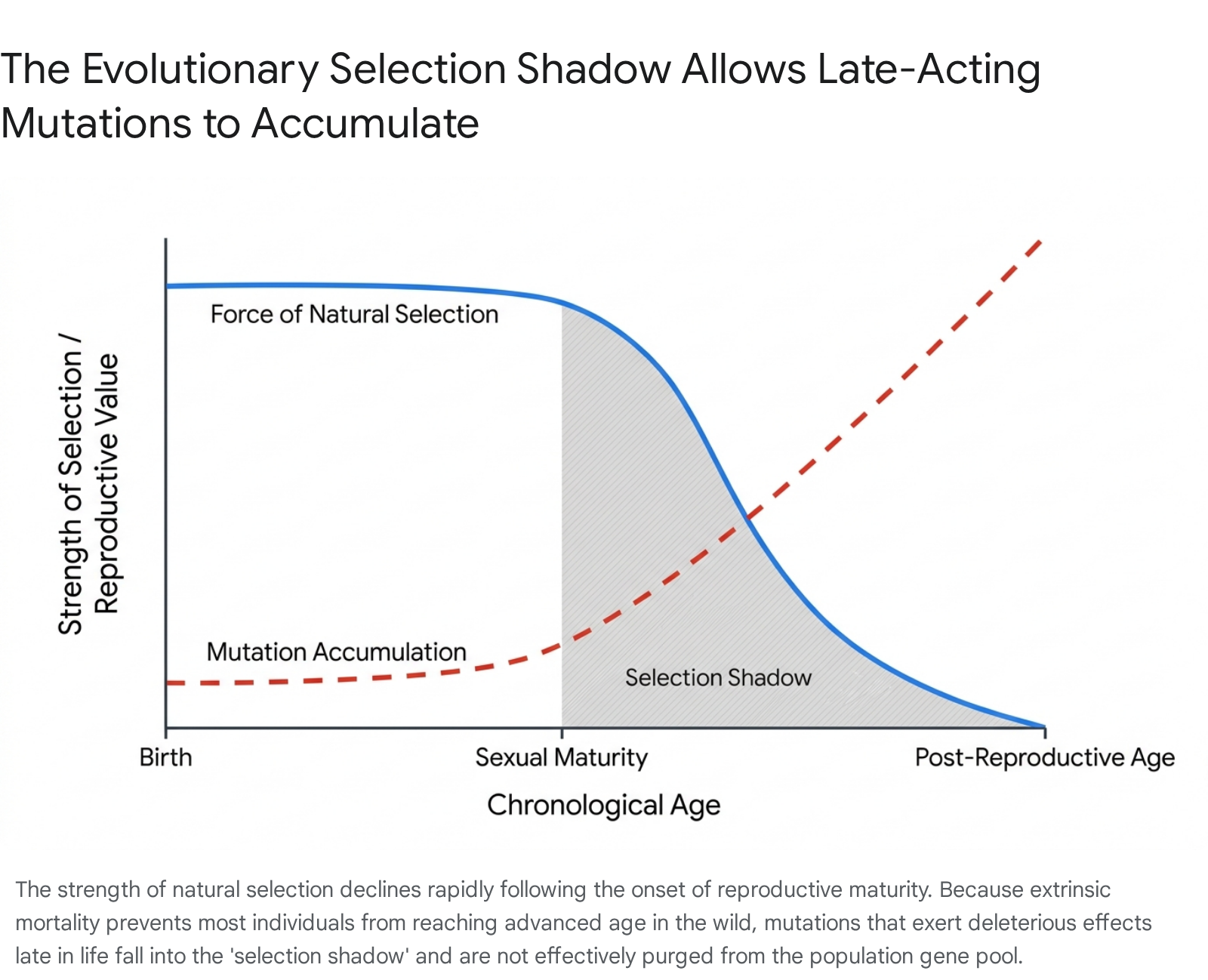
Over evolutionary time, generations of these late-acting deleterious alleles accumulate in the population's gene pool through genetic drift and recurrent mutation pressure 2. When individuals are removed from hazardous wild environments and placed in protected settings (such as laboratories, zoological parks, or modern human societies), they live long enough to experience the compounding pathological effects of this accumulated genetic load. In the Medawar framework, biological aging is simply the revelation of this late-acting mutational load that evolution never bothered to clean up 24.
Mathematical Formalization of Selection Forces
Medawar's hypothesis remained primarily a qualitative narrative until 1966, when evolutionary biologist W.D. Hamilton mathematically formalized the forces of natural selection acting on age-specific survival and fecundity 101112. Hamilton's work provided the definitive mathematical proof that, under standard demographic conditions, the force of natural selection must invariably decline with age.
Hamilton's 1966 Formulations
Hamilton utilized the Euler-Lotka equation, a fundamental theorem in population ecology and demography that defines the intrinsic rate of population increase ($r$) in a stable age distribution 131415:
$$\int_{0}^{\infty} e^{-rx} l_x m_x \,dx = 1$$
In this continuous framework (often applied in discrete form for life-table analysis), $l_x$ represents age-specific survivorship (the probability of surviving from birth to age $x$), and $m_x$ represents age-specific fecundity (the average number of female offspring produced by an individual of age $x$) 131516.
Hamilton asked a fundamental evolutionary question: how would a newly introduced genetic mutation that alters survival at a specific age ($p_a$) affect the organism's total fitness ($r$)? He derived mathematical indicators for the force of selection, demonstrating that the sensitivity of fitness to changes in age-specific survival is directly proportional to the organism's remaining reproductive potential, adjusted for the timing of those offspring 101315. Hamilton's indicator for the force of selection on survival ($s_x$) fundamentally relies on Fisher's concept of reproductive value ($V_x$).
Because $l_x$ is a monotonically decreasing function (survivorship can only drop or remain constant over time; an organism cannot become "un-dead"), and $m_x$ eventually drops to zero in post-reproductive life, the mathematical weighting of any survival or mortality event at age $x$ inevitably declines as $x$ increases 71015. This elegant formalization proved that the decline in the force of natural selection with age is not merely a biological trend, but a mathematical inevitability for any organism with a separation of germ and soma, and a structured life history where reproduction is clustered in earlier life phases 1115. Hamilton concluded that "senescence is an inevitable outcome of evolution" because selection gradients must slope downward across adult lifespan 1012.
Alternative Indicators and Survival Trajectories
While Hamilton's conclusion formed the bedrock of modern evolutionary gerontology, recent mathematical reappraisals have added nuance to his absolute claims. Researchers such as Annette Baudisch have demonstrated that Hamilton's specific indicators of the force of selection implicitly assumed that genetic mutations exert additive effects on mortality 101315.
By applying alternative parameterizations - specifically looking at proportional rather than additive mutational effects on a log scale - Baudisch showed that the mathematical indicators of the force of selection do not necessarily decline continuously 1016. Under certain demographic trajectories where age-specific fecundity ($m_x$) increases sharply with age, the force of selection against deleterious mutations can actually increase during portions of adulthood 101316. This mathematical revelation is critical for explaining species that do not undergo conventional senescence, as it proves the selection shadow is contingent upon specific life-history parameters (namely, early and declining fecundity), rather than an unavoidable physical law of the universe.
Comparative Evolutionary Theories of Senescence
The Mutation Accumulation theory does not exist in isolation. It represents one of three foundational pillars in evolutionary gerontology. Shortly after Medawar introduced MA, George C. Williams proposed the Antagonistic Pleiotropy (AP) theory in 1957 31721. Twenty years later, Thomas Kirkwood introduced the Disposable Soma (DS) theory in 1977 1819. While all three theories rely upon the foundational logic of the selection shadow and Hamilton's declining selection gradients, they invoke fundamentally different physiological and genetic mechanisms.
Antagonistic Pleiotropy
While Mutation Accumulation assumes that aging is caused by a passive accumulation of late-acting mutations that are selectively neutral early in life, Antagonistic Pleiotropy posits that aging is driven by genes that are actively selected for 35. Pleiotropy occurs when a single gene influences multiple, seemingly unrelated phenotypic traits. Williams hypothesized that certain genes confer robust fitness advantages during early development and reproductive maturity, but also manifest severe, detrimental side effects late in life 2120.
Because the force of selection is vastly stronger in youth, a mutation that boosts early fecundity by a mere 5% but causes fatal organ failure at age 70 will easily sweep to fixation in a wild population where few individuals survive past age 40 anyway 42125. Under AP, senescence is an inescapable trade-off directly wired into the genome. A classically cited example involves alleles that promote aggressive calcium deposition for rapid bone growth in youth, which subsequently cause lethal atherosclerotic arterial calcification in old age 4.
A major empirical challenge for AP is the assumption of an unbreakable linkage between the early benefit and the late cost. Standard population genetics theory suggests that over millions of years, selective pressures should eventually decouple these effects through modifier genes, allowing the organism to reap the early benefits without paying the late-life penalty 17212122. Yet, aging persists universally. Consequently, some theorists argue that AP is not a rigid precondition, but an evolved "evolvability adaptation" that protects long-term group-level benefits (like preventing population overshoot and starvation) from being lost to short-term individual selection 1721.
Disposable Soma
The Disposable Soma theory shifts the focus from population genetics to physiological resource allocation and bioenergetics . Kirkwood argued that every organism operates within a finite energy budget. Biological energy must be partitioned between somatic maintenance (e.g., DNA repair, antioxidant defense, proteostasis, immune surveillance) and essential outward functions like growth and reproduction 181928.
In hazardous wild environments with high extrinsic mortality, investing heavily in somatic repair to build a body capable of lasting a century is a severe evolutionary misallocation if the organism is statistically guaranteed to be eaten by a predator or succumb to winter starvation within two years 618. Therefore, natural selection optimizes a deliberate under-investment in somatic maintenance - repairing just enough molecular damage to keep the organism healthy through its expected ecological lifespan. The resulting physiological decay over time, driven by accumulated molecular damage, constitutes senescence 181922.
Theoretical Synergies and Distinctions
The primary distinction between the evolutionary models rests in the nature of the genetic and physiological action. MA is a passive drift of specialized late-acting defects; AP is an active selection of rigid biological trade-offs; and DS represents a systemic, optimized resource deficit.
| Feature | Mutation Accumulation (Medawar) | Antagonistic Pleiotropy (Williams) | Disposable Soma (Kirkwood) |
|---|---|---|---|
| Core Evolutionary Mechanism | Passive genetic drift of late-acting deleterious alleles. | Active natural selection of alleles with early benefits and late costs. | Optimized physiological resource trade-off between reproduction and cellular repair. |
| Role of Natural Selection | Weak selection (Selection Shadow) fails to purge mutations. | Strong early selection actively drives the fixation of the pleiotropic gene. | Selection optimizes energy allocation based on extrinsic mortality risk. |
| Genetic Architecture | Requires a vast polygenic load of highly age-specific, late-acting deleterious alleles. | Requires pleiotropic genes where opposing temporal effects are inextricably linked. | Requires regulatory genes that control energy distribution and basal DNA repair fidelity. |
| Nature of the "Defect" | Distinct, discrete mutations causing specific late-life pathologies. | Systemic design flaws where early-life adaptations organically cause late-life damage. | A generalized, systemic deficit in molecular repair mechanisms leading to cellular entropy. |
| Reversibility / Intervention | Highly heterogeneous and difficult to drug (requires addressing thousands of distinct mutations). | Difficult to reverse without compromising early-life vitality, growth, or reproduction. | Potentially modifiable by altering cellular maintenance pathways (e.g., caloric restriction mimetics). |
Contemporary genomic analyses indicate that these three theories are not mutually exclusive. Modern evolutionary biologists generally accept that aging is a multifaceted phenomenon driven by a combination of MA, AP, and DS mechanisms acting simultaneously across different physiological systems 328.
Somatic Mutation Theory
Medawar's original 1952 formulation of the Mutation Accumulation theory concerned germline mutations accumulating in intergenerational populations over millions of years 2324. However, the same mathematical logic has been extended to the cellular level within a single organism's lifespan. In the "somatic mutation theory of aging," individual somatic cells act as independent evolutionary units. Because somatic cells are not passed on to the next generation, they exist entirely within the selection shadow; any mutations they acquire are "disposable" from an evolutionary standpoint once the organism has successfully reproduced 192425.
For decades, the somatic mutation theory remained an attractive but untestable hypothesis because quantifying stochastic, low-frequency mutations across millions of differentiated cells was technologically impossible due to sequencing noise 24. However, recent advancements in single-cell and high-fidelity whole-genome sequencing have revolutionized the empirical testing of this theory.
Constraints Across Mammalian Species
A landmark 2022 study by Cagan et al. utilized whole-genome sequencing of 208 intestinal crypts across 16 different mammalian species, spanning diverse life histories from mice (lifespan ~3 years) to humans (lifespan ~80 years) and long-lived mammals like giraffes and naked mole rats 262728. The results provided profound empirical support for the constraint of somatic mutation rates by evolutionary pressure.
Cagan et al. demonstrated a striking, robust inverse relationship between a species' somatic mutation rate and its maximal lifespan. Mice accumulate approximately 796 somatic base substitutions per year, while humans accumulate a mere 47 substitutions per year 28. Despite immense disparities in body mass (a 40,000-fold difference between mice and giraffes) and lifespan (an approximately 30-fold difference), the absolute end-of-lifespan mutational burden was remarkably conserved. Mammals appear to reach the end of their natural lifespans with a similar total accumulation of somatic mutations (a variation of only roughly 3-fold across species) 26272829.
The mutational signatures isolated in these diverse species were broadly similar, predominantly driven by endogenous cellular processes such as oxidative damage and 5-methylcytosine deamination 262728. This implies that the source of the genomic damage is universal, dictated by the basic chemistry of carbon-based life, but the efficiency of the DNA repair mechanisms - dictated by the evolutionary allocation of resources predicted by the Disposable Soma theory - varies precisely based on the species' ecological niche.
Peto's Paradox and Cancer Resistance
The scaling of somatic mutation rates with lifespan provides critical insight into Peto's Paradox. Simple mathematical models of carcinogenesis dictate that organisms with vastly more cells (greater body mass) and longer lifespans should have an exponentially higher risk of developing cancer, as each cell division carries a baseline risk of oncogenic mutation. However, empirical observation shows that large, long-lived animals (like whales and elephants) do not exhibit higher cancer rates than small, short-lived animals (like mice) 3031.
The genomic data suggests that evolution resolves Peto's paradox not merely through specialized tumor suppressor duplication (though this occurs in some species like elephants), but by universally dialing down the basal somatic mutation rate in long-lived species 2631. Because larger animals naturally suffer lower extrinsic mortality (they possess fewer natural predators), the selection shadow is pushed further back in chronological time. This prolonged reproductive window creates a strong selective pressure to invest more heavily in DNA repair mechanisms, drastically slowing the rate of somatic mutation accumulation 3031. The positive feedback loop forces the evolution of higher fidelity repair, effectively delaying the onset of both cancer and general senescence 30.
Germline and Somatic Mutation Dynamics
A nuanced understanding of the Mutation Accumulation theory requires delineating the Weismann barrier - the strict biological separation between the potentially immortal germline lineage (sperm and egg cells) and the mortal somatic cells 919.
The Weismann Barrier and Lineage Protection
If DNA damage and mutation accumulation are unavoidable thermodynamic realities of biological chemistry, why do species not degenerate over successive generations? The answer lies in the intense evolutionary pressure to protect the germline. Natural selection maintains the fidelity of germ cells with extreme prejudice, as any defects here directly jeopardize the lineage's survival and evolutionary continuity 1932. Consequently, germ cells benefit from highly upregulated DNA repair pathways, specialized chromatin configurations, and rigorous apoptotic clearing mechanisms that rapidly eliminate damaged cells.
Recent sequencing technologies have confirmed this stark dichotomy. The mutation rate in somatic cells is estimated to be one to two orders of magnitude higher than the mutation rate in germ cells 2433. A single human sperm cell typically contains fewer than 100 de novo mutations, whereas differentiated human somatic cells, such as fibroblasts or intestinal epithelial cells, easily accumulate thousands of base substitutions and structural variations over a standard lifespan 24.
Paternal Age and De Novo Mutations
Interestingly, while the somatic mutation rate drives the physiological aging of the individual, the slowly ticking germline mutation rate is highly sensitive to the chronological age of the parent. Advanced paternal age correlates heavily with an exponentially increased burden of de novo mutations passed to offspring, increasing the risk of developmental and neurological disorders 33. This trend highlights that even the highly protected germline is not entirely immune to the physics of time and cumulative cellular divisions. The interplay between these two rates reflects the ultimate manifestation of the Disposable Soma theory: the soma is permitted to mutate and decay because it is an evolutionary dead end, while metabolic energy is disproportionately allocated to shielding the germline genome 1932.
Advanced Sequencing and Clonal Expansion
While early evolutionary debates viewed the Mutation Accumulation theory strictly through the lens of population genetics, modern multi-omics integration has mapped these evolutionary abstractions directly onto human pathology. Contemporary sequencing capabilities reveal that somatic mutation accumulation is not merely an abstract concept, but a tangible, chaotic disruption of the cellular landscape that actively precipitates age-related disease.
High-Fidelity Sequencing Techniques
The historical barrier to proving the somatic mutation theory has always been the baseline error rate of sequencing machinery, which struggles to differentiate a true biological mutation from a polymerase sequencing artifact. In 2024, the development of Hairpin Duplex Enhanced Fidelity Sequencing (HiDEF-seq) allowed researchers to detect molecular changes in DNA before they even become permanent double-strand mutations 34. By accurately reading single-strand base changes with an astonishingly low error rate of an estimated one recording error per 100 trillion base pairs, techniques like HiDEF-seq provide a highly granular view of how intrinsic chemical decay (such as cytosine deamination) initiates the mutation accumulation cascade in completely healthy cells long before carcinogenesis 34.
Clonal Hematopoiesis and Systemic Pathology
One of the most profound clinical validations of the somatic MA framework in recent years has been the discovery of Clonal Hematopoiesis of Indeterminate Potential (CHIP). As humans age, the hematopoietic stem cell (HSC) pool in the bone marrow accumulates somatic mutations. While most are neutral passenger mutations, some grant a competitive survival advantage to specific stem cells 243135.
A mutant HSC will undergo clonal expansion, rapidly populating the blood compartment with its mutant progeny. While these expansions are frequently driven by known cancer-associated genes, CHIP routinely occurs in healthy individuals without leukemia. Crucially, this accumulation of mutant clones correlates heavily with severe late-onset pathologies that are seemingly unrelated to the blood compartment, particularly cardiovascular disease and atherosclerosis, driven by a hyper-inflammatory phenotype in the mutant macrophages 243135.
This phenomenon embodies the exact hazard predicted by the interaction between MA and DS theories. Because humans evolved in environments where life expectancy rarely exceeded 40 years, there was minimal selective pressure to evolve foolproof suppression of slow-growing, mildly advantageous stem cell clones. The mutational drift is effectively silent during youth but devastates the cardiovascular system in the extended lifespan of modern society. Recent high-impact analyses of the UK Biobank involving over 200,000 individuals have vastly expanded the pool of genes implicated in these clonal expansions, uncovering 17 newly discovered driver genes and reinforcing that age-related mutation accumulation drives systemic functional decline long before it initiates oncogenesis 35.
Somatic Mutations versus Epigenetic Clocks
Despite these technical triumphs, a deep schism remains in biogerontology regarding whether somatic mutations are the primary driver of aging, or merely a downstream symptom of a deeper decay. The rise of "epigenetic clocks" - which track the predictable, age-related methylation drift of CpG islands - has challenged the primacy of DNA sequence mutations 825. While mutations are stochastic, sporadic, and highly variable between adjacent cells, epigenetic drift is remarkably consistent across individuals and tissues.
However, recent integrative models suggest a biological reconciliation. The stochastic accumulation of somatic DNA mutations frequently forces surrounding genomic regions to undergo epigenetic remodeling to maintain stability. Studies matching individual somatic mutation profiles against their epigenetic clocks indicate that specific DNA mutations correlate tightly with broad, predictable methylation shifts in the surrounding DNA 25. Furthermore, somatic mutation profiles can explain almost 50% of the variation in methylation age across individuals 25. This implies that the highly variable, random accumulation of mutations predicted by Medawar effectively serves as the underlying biological "entropy" that drives the more predictable, systemic failure of epigenetic maintenance over time 25.
Empirical Challenges to Mutation Accumulation
While the selection shadow and declining selection gradients provide an elegant framework for most mammalian aging, the Mutation Accumulation theory faces severe empirical and theoretical challenges when applied across the broader tree of life. If Hamilton's declining selection forces represent an absolute mathematical inevitability, senescence should be a universal characteristic of multicellular life 71015. However, robust demographic data increasingly demonstrate that this is not the case.
Negligible Senescence and Indeterminate Growth
Several species display what biogerontologist Caleb Finch termed "negligible senescence" - a complete absence of measurable functional decline, reproductive senescence, or increasing mortality rates with chronological age past maturity 363738. In these organisms, the Gompertz-Makeham law of mortality (which dictates that mortality risk grows exponentially with age) breaks down entirely 37.
| Species | Common Name | Maximum Documented Lifespan | Senescence Profile |
|---|---|---|---|
| Heterocephalus glaber | Naked Mole Rat | ~30+ years | Defies Gompertz-Makeham law; no mortality increase with age; highly efficient cellular repair relative to body mass. |
| Arctica islandica | Ocean Quahog | >500 years | Negligible senescence; exceptional resistance to oxidative stress and precise proteostasis. |
| Sebastes aleutianus | Rougheye Rockfish | >200 years | Indeterminate growth; constant or declining mortality with adult age. |
| Pinus longaeva | Great Basin Bristlecone Pine | >5,000 years | Modular organism; persistent stem cell meristems; no demographic evidence of physiological senescence. |
Note: Lifespan estimates and senescence parameters derived from multiple genomic databases and empirical life-history tracking 36373839.
How can the Mutation Accumulation theory account for species that demonstrably do not age? The key lies in revisiting Hamilton's underlying demographic assumptions. Hamilton implicitly assumed a life history where fecundity remains relatively stable or peaks early, and growth ceases at maturity (determinate growth) 712. However, many species of fish (like the rougheye rockfish), reptiles, and basal metazoans (like Hydra) exhibit indeterminate growth - they continue to grow larger throughout their entire lifespans.
In species with indeterminate growth, larger body size often translates to exponentially higher fecundity and a reduced risk of predation (lower extrinsic mortality) 640. Therefore, the reproductive value ($V_x$) of these organisms actually increases with chronological age. If older individuals contribute disproportionately more offspring to the next generation than younger individuals, the selection shadow never manifests. The force of natural selection remains persistently high, ruthlessly purging deleterious late-acting mutations and maintaining robust somatic repair mechanisms indefinitely 4630. Thus, negligible senescence does not disprove the MA theory; rather, it proves that MA correctly predicts aging only when ecological parameters force a decline in reproductive value.
Late-Life Mortality Deceleration
Another significant challenge to the classic MA and AP models is the behavior of mortality rates at extreme old ages. Classic theories predict a "wall of death" - a rapid, terminal spike in mortality once a cohort passes its maximum evolutionary lifespan, as the genome becomes entirely saturated with late-acting deleterious alleles 2341.
However, large-scale biodemographic studies of human populations and insect models (such as Drosophila melanogaster) have consistently revealed a phenomenon known as "late-life mortality deceleration" 53748. At extremely advanced ages (e.g., human centenarians), the exponential increase in mortality risk flattens out into a demographic plateau. If aging is exclusively driven by an uncontrolled accumulation of unselected deleterious alleles, the mortality curve should theoretically approach 100% sharply, rather than plateauing 2. Some theorists resolve this discrepancy by arguing for demographic heterogeneity - frail individuals die off early, leaving only highly genetically robust individuals in the oldest cohorts - while others suggest that physiological systems reach a state of terminal redundancy exhaustion, where damage is maximal but mechanically stable 548.
Post-Reproductive Survival and Inclusive Fitness
Furthermore, the existence of prolonged post-reproductive lifespan in humans and some toothed whales contradicts strict formulations of the selection shadow. If reproduction ceases at menopause, the direct reproductive value is zero, and the organism should theoretically succumb to mutation accumulation immediately 41.
Modern evolutionary frameworks suggest that indirect fitness contributions exert a secondary force of selection that maintains somatic integrity well past the cessation of direct fertility. The "Grandmother Hypothesis" posits that in highly social species, older individuals provision resources, transfer complex foraging skills, and enhance the survival of kin 184142. By actively improving the survival probabilities of their grand-offspring, post-reproductive individuals increase their inclusive fitness. The evolutionary mathematics of inclusive fitness (often modeled via Hamilton's separate rule for kin selection) effectively extends the selection shadow outward, continuing to select against late-acting deleterious mutations to preserve the physical and cognitive capacity necessary for intergenerational resource transfer 4142.
Demographic Shifts and the Future of Mutation Accumulation
The parameters that define the selection shadow are not fixed; they are highly dependent on the external environment. Recent studies leveraging massive demographic datasets from 175 countries spanning from 1950 to 2023 indicate that the modern human environment is radically altering the evolutionary forces acting upon our species 50.
Post-Transition Population Dynamics
In modern, post-demographic-transition populations, massive reductions in infant mortality, infectious disease, and starvation have virtually eliminated the traditional sources of extrinsic mortality. A 2025 analysis of these demographic schedules revealed a stark "extension-dilution trade-off." The age at which selection intensity halves has increased by nearly 1.7 years since 1950, extending the reach of natural selection deeper into chronological age 50.
However, paradoxically, the peak intensity of selection has declined by nearly 30% 50. Because almost everyone survives to reproductive age regardless of their underlying genetic robustness, absolute selection at all ages has weakened. Late-acting deleterious mutations face vastly reduced purifying pressure. The ratio of selection intensity at age 20 to intensity at age 40 has risen substantially, steepening the gradient that favors alleles with early benefits over late-life costs 50. Therefore, while medical interventions continually extend human lifespan, the fundamental genetic potential for both Mutation Accumulation and Antagonistic Pleiotropy to propagate through modern human populations is actually being amplified by our own demographic success 50.
By leveraging the inescapable mathematics of the selection shadow, Peter Medawar provided a foundational mechanism for why biological forms inevitably deteriorate, shifting the paradigm of senescence from a programmed necessity to a structural vulnerability inherent to life histories. The recent revolution in genomic sequencing has breathed new life into the somatic corollary of the theory, demonstrating that mutation rates are exquisitely tuned across the mammalian class to inversely correlate with species lifespan, directly validating the concept that evolutionary selective pressure governs the velocity of genetic decay.