Molecular mechanisms and therapeutics of intrinsic cardiac aging
1. Delineating Intrinsic Biological Cardiac Aging from Secondary Cardiovascular Disease
The prevailing paradigm of cardiovascular medicine has traditionally conflated chronological aging with the cumulative burden of secondary cardiovascular risk factors. Under this conventional framework, age-related cardiac dysfunction is frequently and mistakenly attributed entirely to the lifelong mechanical and metabolic wear-and-tear wrought by secondary pathologies, such as atherosclerosis, chronic systemic hypertension, ischemic heart disease, and diabetes mellitus. However, contemporary geroscience necessitates a strict and fundamental demarcation: intrinsic biological cardiac aging is a distinct, cell-autonomous, and evolutionarily programmed decay that occurs entirely independently of systemic secondary diseases 12.
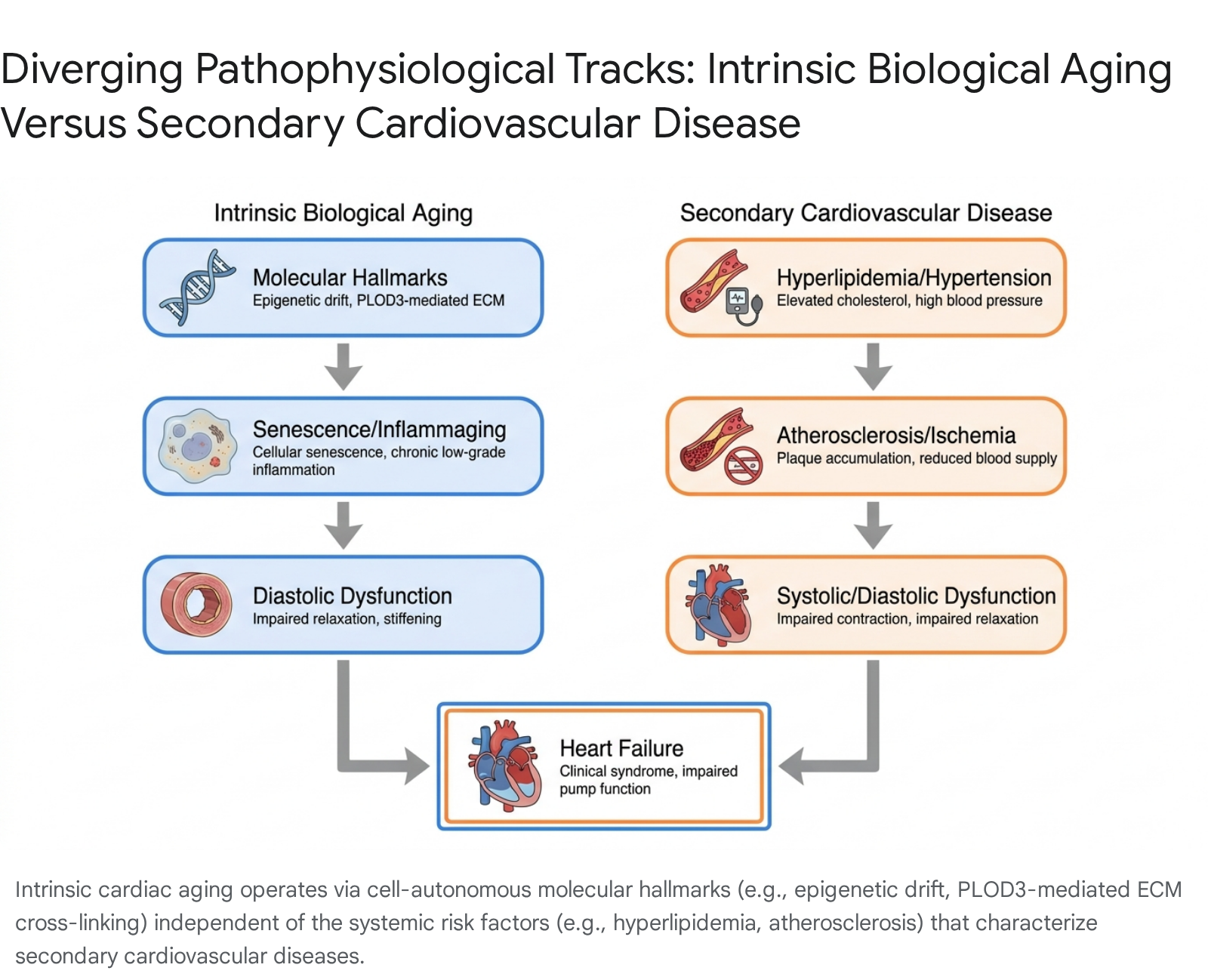
In the complete absence of coronary artery occlusion, systemic afterload elevations, or metabolic syndrome, the myocardium undergoes profound, inherent structural and functional transformations as chronological time advances. Intrinsic cardiac aging manifests macroscopically as left ventricular concentric hypertrophy, heightened myocardial passive stiffness, reduced cardiac reserve, and isolated diastolic dysfunction 13. At the histological and cellular levels, it is characterized by extensive cardiomyocyte hypertrophy, interstitial fibrosis, and profound vascular rarefaction 12. These structural shifts are not merely the fibrotic scars of prior ischemic insults but are the downstream pathophysiological consequences of fundamental biological aging hallmarks, including telomere attrition, genomic instability, epigenetic drift, and the progressive loss of proteostasis 345.
Failing to isolate intrinsic biological aging from secondary cardiovascular disease severely limits the conceptualization and development of disease-modifying therapies. Interventions targeting low-density lipoprotein cholesterol or systemic blood pressure - while undeniably critical for managing atherosclerosis and hypertension - do not halt the intrinsic biological clock of the cardiomyocyte, the cardiac fibroblast, or the endothelial cell. Consequently, a new frontier in clinical cardiology has emerged, focusing on directly targeting the molecular hallmarks of biological senescence to extend cardiovascular healthspan and specifically address recalcitrant pathologies like Heart Failure with Preserved Ejection Fraction (HFpEF), which is intrinsically linked to biological aging 567.
To formalize the assessment of these intrinsic aging processes, international scientific bodies, including the Aging Biomarker Consortium (ABC) of China, have recently published comprehensive consensus frameworks to standardize the measurement of cardiac aging across diverse populations 8910. This 2023 - 2024 consensus establishes a highly structured methodology for characterizing aging biomarkers across three distinct dimensions. The functional dimension encompasses a broad spectrum of markers reflecting diastolic and systolic reserve, sinus node pacing, neuroendocrine secretion, and coronary microcirculation 8. The structural domain emphasizes advanced imaging markers relevant to concentric cardiac remodeling and epicardial fat deposition, while the humoral aspect underscores both systemic and heart-specific markers, including circulating cytokines and plasma metabolites 811. By isolating these dimensions from classical disease biomarkers, researchers can accurately assess the biological functional status of the heart and predict the risk of age-related failure independent of generic atherosclerotic burden 910.
2. Molecular Drivers of Intrinsic Cardiac Senescence
The contemporary framework of biological aging has expanded significantly to encompass twelve highly interconnected hallmarks, incorporating processes such as impaired autophagy, chronic inflammation, and dysbiosis alongside traditional genomic and mitochondrial instability 512. In the context of the myocardium, high-impact research post-2023 has definitively broadened the mechanistic focus beyond simple theories of oxidative stress and calcium mishandling. Current investigations heavily emphasize the dynamic regulation of extracellular matrix remodeling, sophisticated epigenetic reprogramming, and the systemic, tissue-degrading impact of chronic low-grade inflammation.
2.1 Extracellular Matrix Remodeling: Beyond Simple Collagen Accumulation
Historically, age-related myocardial stiffness was attributed to a simple, linear net overproduction and accumulation of fibrillar collagens (specifically types I and III) driven by continuously active cardiac fibroblasts. However, high-resolution mass spectrometry and advanced proteomic network analyses have fundamentally challenged this foundational assumption. Recent meticulous evaluations of the murine atrial and ventricular extracellular matrix reveal that the overall absolute collagen content may actually decrease with advanced chronological age, contrasting sharply with the traditional fibrosis model 13.
Instead, the primary driver of age-associated myocardial stiffness and mechanical failure is a profound alteration in collagen architecture, specifically driven by enhanced enzymatic and non-enzymatic cross-linking 131415. Advanced glycation end-products accumulate continuously on long-lived extracellular matrix proteins, forming highly stable, irreversible cross-links that resist normal matrix metalloproteinase degradation, thereby locking the matrix into a rigid conformation 1314. Concurrently, enzymatic cross-linking is heavily upregulated during biological aging. Stringent network mappings of physical protein associations have identified Procollagen-Lysine,2-Oxoglutarate 5-Dioxygenase 3 (PLOD3) and various Lysyl Oxidase isoforms as critical mediators that facilitate excessive, pathological collagen cross-linking in the aging heart, fundamentally altering the mechanical properties of the tissue without necessarily increasing total collagen mass 1317.
Furthermore, the aging extracellular matrix is not merely a passive structural scaffold; it functions as an active, pro-fibrotic signaling entity capable of overriding cellular youth. Innovative engineered hybrid hydrogel systems utilizing decellularized matrix from aged murine hearts have demonstrated that this aged matrix autonomously drives young, healthy cardiac fibroblasts into a pathogenic myofibroblast transition 1517. This illustrates that age-related alterations in matrix mechanics and specific ligand presentations - such as increased matrix fibronectin and altered integrin signaling - directly dictate cellular senescence and dysfunction, overriding the intrinsic biological age of the resident cells 1517. The aging matrix also exhibits distinct sexual dimorphism; post-menopausal females lose the protective effect of estrogen, which typically suppresses excessive matrix remodeling, leading to a disproportionate acceleration in fibrotic accumulation and matrix stiffening compared to age-matched males 15.
2.2 Epigenetic Shifts and the Primate SIRT2-STAT3-CDKN2B Axis
Epigenetic dysregulation - encompassing vast alterations in DNA methylation patterns, histone modification, and chromatin remodeling - is a primary hallmark driving intrinsic cardiac aging 316. While the sirtuin family of NAD+-dependent deacetylases has long been implicated broadly in longevity and metabolic regulation, recent highly targeted investigations have isolated the specific and critical role of Sirtuin 2 (SIRT2) in primate cardiac aging 191718.
Comprehensive transcriptomic and proteomic mappings of young versus naturally aged cynomolgus monkey hearts identified a precipitous, age-dependent decline in SIRT2 expression as a defining molecular feature of the aged primate myocardium 1719. To validate this finding, researchers utilized advanced CRISPR-Cas9 engineering to create SIRT2-deficient human pluripotent stem cell-derived cardiomyocytes. These engineered cells unequivocally demonstrated that SIRT2 deficiency alone is sufficient to recapitulate the cardinal features of primate cardiac aging in vitro, precipitating accelerated cellular senescence, abnormal cellular hypertrophy, and diminished replicative capacity 1719.
The underlying molecular mechanism driving this decay involves the Signal Transducer and Activator of Transcription 3 (STAT3). Under youthful, homeostatic conditions, SIRT2 directly interacts with and deacetylates STAT3 at lysine 685, maintaining it in a tightly regulated state 19. As SIRT2 levels fall during biological aging, STAT3 becomes chronically hyperacetylated, which hyperactivates its transcriptional activity. This leads directly to the robust upregulation of the cyclin-dependent kinase inhibitor CDKN2B, which permanently arrests the cell cycle and drives the cardiomyocyte into a terminal, senescent, and highly dysfunctional state 1719. In vivo validation has shown that AAV-mediated intra-myocardial delivery of SIRT2 effectively ameliorates age-related cardiac dysfunction in murine models, establishing the SIRT2-STAT3-CDKN2B epigenetic axis as a highly viable target for clinical rejuvenation 191723. The integration of spatial transcriptomics has further illuminated this epigenetic landscape, revealing that such transcriptional changes are not uniform but occur with distinct spatial heterogeneity across different myocardial compartments during the aging trajectory 20.
2.3 Inflammaging, cGAS-STING, and the Senescence-Associated Secretory Phenotype
Cellular senescence within the cardiovascular system is characterized by a permanent state of cell-cycle arrest coupled with profound metabolic and secretory shifts 56. A critical consequence of this state is the Senescence-Associated Secretory Phenotype (SASP). Unlike embryonic senescence, which relies heavily on growth factors to orchestrate developmental tissue remodeling, postnatal cellular senescence is highly pathogenic 6. Senescent cardiomyocytes, endothelial cells, and cardiac fibroblasts continuously secrete a toxic cocktail of pro-inflammatory cytokines (including interleukin-1 beta, interleukin-6, and tumor necrosis factor-alpha), destructive chemokines, and aberrant matrix-degrading proteases 56.
This persistent, low-grade, sterile systemic and localized inflammation is termed "inflammaging." Unlike acute inflammatory responses to injury or infection, inflammaging arises without overt pathogenic triggers and represents a sustained, destructive equilibrium 5. Inflammaging operates via multiple interconnected pathways, primarily driven by the cytosolic DNA-sensing cGAS-STING pathway 21. In aged cardiomyocytes, cumulative mitochondrial dysfunction and genomic instability result in the leakage of mitochondrial and nuclear DNA into the cytosol. The cGAS enzyme detects this misplaced DNA and synthesizes the second messenger cyclic GMP-AMP, which activates STING. This cascade triggers the recruitment and phosphorylation of TBK1, leading to the subsequent activation of the transcription factors NF-kappaB and IRF3, which sustain the massive transcription of SASP inflammatory cytokines 1021. This chronic inflammatory milieu acts via autocrine and paracrine signaling to spread senescence to neighboring healthy cells, accelerating global myocardial functional decline, fundamentally impairing nitric oxide bioavailability, and fostering a rigid, pro-fibrotic environment highly conducive to the pathogenesis of diastolic heart failure 52223.
2.4 Metabolic Inflexibility, Mitochondrial Dysfunction, and Calcium Mishandling
Intrinsic cardiac aging is inextricably tied to profound metabolic inflexibility and structural mitochondrial decay. The youthful heart is a metabolic omnivore, relying heavily on efficient fatty acid oxidation to meet its massive energetic demands. However, the aging heart exhibits a reduced capacity for fatty acid oxidation and an increased, maladaptive reliance on less efficient glucose metabolism 2324. This metabolic shift is intimately connected to the dysregulation of primary nutrient-sensing pathways, particularly the mechanistic target of rapamycin and AMP-activated protein kinase 524.
Concurrent, precipitous declines in baseline autophagy and specialized mitophagy lead to the progressive accumulation of fragmented, massive reactive oxygen species-generating mitochondria 2324. This severe cellular energy deficit directly impairs highly ATP-dependent cellular processes, most notably calcium handling during the cardiac cycle. The Sarcoplasmic/Endoplasmic Reticulum Calcium ATPase 2a (SERCA2a), the essential pump responsible for rapidly re-sequestering calcium into the sarcoplasmic reticulum to facilitate diastole, is highly sensitive to both oxidative damage and ATP depletion 12530. The age-dependent downregulation and functional impairment of SERCA2a directly result in chronically elevated resting cytosolic calcium levels, drastically prolonged myocardial relaxation times, and the classic diastolic dysfunction that serves as the mechanical hallmark of the aged heart 25.
Table 1: Structural Hallmarks of Cardiac Aging and Underlying Molecular Drivers
| Structural Hallmark | Cellular / Molecular Driver | Pathophysiological Consequence |
|---|---|---|
| Increased Myocardial Stiffness | Upregulation of PLOD3 and LOX enzymes; excessive accumulation of AGEs on ECM proteins. | Hyper-cross-linking of collagen network; increased resistance to physiological MMP degradation; impaired ventricular filling mechanics. |
| Cardiomyocyte Hypertrophy | Precipitous downregulation of SIRT2; subsequent hyperacetylation of STAT3; upregulation of CDKN2B. | Compensatory cellular enlargement; rapid progression toward permanent cellular senescence and functional exhaustion. |
| Diastolic Dysfunction | Severe oxidative damage to SERCA2a; profound mitochondrial ATP deficits; globally impaired autophagy/mitophagy. | Reduced rate of active calcium reuptake into the sarcoplasmic reticulum; critically delayed myocardial relaxation; elevated filling pressures. |
| Interstitial Fibrosis | SASP-mediated secretion of TGF-beta; intense cGAS-STING pathway activation via cytosolic DNA sensing. | Paracrine activation of quiescent cardiac fibroblasts into active, matrix-depositing myofibroblasts; chronic low-grade sterile inflammation. |
| Vascular Rarefaction | Critical telomere attrition in endothelial cells; decreased endothelial nitric oxide bioavailability; eNOS/nNOS dysfunction. | Premature endothelial senescence; severely impaired coronary microcirculation; heightened arterial stiffness and impaired vasodilation. |
3. The Epidemiological Link Between Biological Aging and HFpEF: A Global Perspective
Heart Failure with Preserved Ejection Fraction (HFpEF) is widely recognized as the ultimate macroscopic, clinical manifestation of intrinsic cardiac aging. As the global population ages at an unprecedented rate, HFpEF incidence is rising exponentially, now accounting for over 50% of all heart failure cases worldwide, with its prevalence predicted to continue increasing at a rate of 1% per year relative to heart failure with reduced ejection fraction 3126. However, the long-held assumption that HFpEF is a monolithic disease exclusively afflicting elderly, obese Western populations has been fundamentally dismantled by recent global epidemiological data from geographically diverse and multi-ethnic cohorts. Biological aging does not progress at a uniform chronological rate globally; it is heavily modified by regional epigenetic, environmental, and ethnic factors.
3.1 Divergent Phenotypes Across Asian Cohorts
Registries relying exclusively on North American or Western European data frequently describe the prototypical HFpEF patient as an elderly female (often well over 75 years of age) with multiple metabolic comorbidities, notably severe obesity, long-standing atrial fibrillation, and systemic metabolic syndrome 3334. However, active clinical surveillance of non-Western populations reveals starkly different trajectories of biological aging and HFpEF manifestation.
Data from the ASIAN-HF (Asian Sudden Cardiac Death in Heart Failure) registry, a massive prospective longitudinal multinational study spanning 11 regions across Asia, demonstrates profound phenotypic heterogeneity that challenges Western norms 27. When analyzing over 1,200 HFpEF patients, researchers found that patients from Southeast Asia exhibit the highest global burden of severe comorbidities, such as end-stage chronic kidney disease and advanced diabetes mellitus. Yet, paradoxically, these patients present with symptomatic HFpEF at a significantly younger chronological age compared to their counterparts in Northeast Asia or Western cohorts 3427. Notably, up to 15% of HFpEF patients in certain Asian demographic subsets are classified clinically as "very young" (under 55 years of age) 34. Despite their younger chronological age, these patients exhibit physiological markers of highly advanced biological aging - such as severe myocardial concentric remodeling and drastically impaired quality of life - driven heavily by the synergistic, destructive effects of lean diabetes and premature vascular aging 27. The presence of concurrent diabetes and chronic kidney disease in these younger Asian cohorts was associated with a more than two-fold higher adjusted hazard of mortality or heart failure hospitalization, indicating an extreme acceleration of the biological aging clock 27.
3.2 Accelerated Biological Aging in Africa and Latin America
Similarly, the INTER-CHF prospective cohort study (enrolling 5,813 patients across Africa, Asia, the Middle East, and South America) and the ongoing THESUS-HF II registry provide critical data that further dismantle Western-centric models of cardiac aging. In sub-Saharan Africa, the onset of clinical heart failure occurs up to a full decade earlier than in Western Europe or North America, with patients frequently presenting in advanced New York Heart Association (NYHA) functional class IV 362829. In these African cohorts, both HFpEF and overall heart failure are predominantly driven by severe, unmitigated hypertensive heart disease, contrasting sharply with the ischemic or obesity-driven etiologies dominant in the West 3629.
Despite presenting at a much younger chronological age, patients in Africa and India suffer the highest one-year mortality rates (34% and 23%, respectively) globally, far exceeding the 7-9% mortality rates observed in China, South America, and the Middle East, and vastly outpacing Western European mortality 2939. This severe, lethal disease manifestation at a younger age suggests an extreme, accelerated pace of biological aging and target-organ damage, compounded heavily by socioeconomic determinants, delayed clinical diagnosis, lower literacy levels, and chronically poor access to disease-modifying therapies, such as the severe underutilization of beta-blockers in African cohorts 362839.
In Latin America, specifically within the PESA study and massive Brazilian longitudinal cohorts, HFpEF epidemiology reveals the highest global proportion of female patients, deeply correlated with widespread, poorly controlled chronic hypertension 3334. The intersection of intrinsic aging and secondary disease is being meticulously tracked in the ELSA-Brasil study, which utilized the Klemera-Doubal method to compute the biological age of over 12,000 Brazilian adults using multisystem biomarkers 30. The results proved that the discrepancy between biological age and chronological age ($\Delta$age) is a potent, independent predictor of cardiovascular mortality, far superseding chronological age alone as a risk metric 30. Furthermore, research into the immense genetic diversity and high degree of genetic admixture in Brazilian supercentenarians is yielding new biomarkers of resilience, such as elevated neutralizing antibodies and plasma Neurofilament light chain parameters, offering profound insights into how certain populations successfully evade rapid cardiovascular senescence despite socioeconomic hardships 313243.
4. The Translational Gap: Model Organisms versus Human Physiology
The translation of geroscience from short-lived model organisms (such as inbred mice or nematodes) to complex human physiology has proven to be an immense and frequently frustrating challenge. Interventions that reliably extend lifespan and beautifully reverse cardiac aging in rodents frequently encounter highly complex physiological barriers, unexpected biphasic dose-responses, and severe unintended systemic side effects when applied to human biology 12. The critical requirement for lengthy, time-consuming clinical studies in humans further exacerbates this translational delay 12. Two prominent examples - the mTOR inhibitor rapamycin and Growth Differentiation Factor 11 - perfectly illustrate this severe translational gap.
4.1 The Rapamycin Paradigm and Partial Cellular Reprogramming
Rapamycin, an inhibitor of the mechanistic target of rapamycin complex 1 (mTORC1), remains one of the most robust and widely cited lifespan-extending pharmacological agents in murine models. In highly aged 22- to 24-month-old mice, short-term (8 to 10 weeks) rapamycin administration yields dramatic, highly measurable reversals in intrinsic cardiac aging. It significantly reduces pathological left ventricular hypertrophy, drastically decreases passive myocardial stiffness, and beautifully restores youthful diastolic function 33. Crucially, proteomic and metabolomic studies reveal that these profound cardiac benefits are highly persistent, remaining robustly detectable up to 8 full weeks after the complete cessation of treatment, driven by enduring changes in the cardiac proteome and persistently altered mitochondrial respiratory chain activity 33.
However, the continuous, systemic administration of rapamycin in humans to achieve anti-aging effects is clinically untenable. Chronic exposure leads to unavoidable mTORC2 inhibition over time, which induces profound systemic insulin resistance, severe dyslipidemia, and dangerous immunosuppression 3334. While the murine data suggesting that the cardiac benefits are "persistent" has prompted researchers to advocate for intermittent, short-term cyclic dosing regimens in humans to capture the anti-aging benefits while mitigating metabolic toxicity, defining the optimal therapeutic window, dosage, and frequency for human cardiac rejuvenation remains a highly complex hurdle that has yet to be solved in large-scale clinical trials 33.
In an attempt to bypass pharmacological toxicity, researchers have explored genetic partial cellular reprogramming using the Yamanaka factors (OSKM). While OSKM expression can reverse epigenetic aging clocks and restore cellular function, its in vivo delivery in humans presents unacceptable risks, specifically the induction of dangerous pluripotency pathways resulting in rampant teratoma formation 3446. This translational barrier has led to the recent screening of novel, non-pluripotent single-gene interventions, such as SB000, which have demonstrated the ability to decouple age reversal from pluripotency, successfully driving multi-omic rejuvenation and decreasing single-cell transcriptomic age in human fibroblasts without causing a loss of somatic identity or function 46. While promising, these genetic reprogramming techniques remain years away from systemic human cardiovascular application.
4.2 The GDF11 Controversy: Evolution of a Scientific Consensus
The turbulent trajectory of Growth Differentiation Factor 11 (GDF11) serves as the ultimate cautionary tale regarding the immense complexities and potential dangers of translational geroscience. In 2013, highly publicized heterochronic parabiosis experiments - which surgically connected the circulatory systems of young and old mice - demonstrated that circulating factors in young blood could remarkably reverse age-related cardiac hypertrophy in the older parabiont after just four weeks of shared circulation 3536. Using modified aptamer-based proteomics, researchers identified GDF11, an obscure member of the TGF-beta superfamily, as the putative "youth factor," claiming its systemic levels steadily declined with age and that exogenous recombinant GDF11 could independently and completely reverse cardiac aging in old mice 3536.
This initial, explosive discovery faced severe, immediate scientific scrutiny. By 2015, multiple independent, highly respected laboratories reported a complete failure to reproduce these anti-hypertrophic findings at the suggested doses. Critical methodological flaws were uncovered, most notably that the commercial antibodies (specifically from Abcam) originally used to detect and quantify GDF11 cross-reacted heavily with myostatin (GDF8), a highly homologous protein that genuinely regulates skeletal muscle mass 3738. Rigorous subsequent studies using highly specific detection methods demonstrated that restoring GDF11 to so-called "youthful" levels had absolutely no positive effect on cardiac structure, myocyte size, or pump function 38.
Post-2022 research has decisively resolved this controversy, establishing a new, nuanced consensus: the physiological effects of GDF11 are highly dose-dependent, biphasic, and fundamentally context-specific 3739. While very low, precise doses of recombinant GDF11 preparations may offer marginal anti-hypertrophic effects in specific pressure-overload murine models, supra-physiological doses (e.g., 5.0 mg/kg) trigger severe, irreversible skeletal muscle cachexia, massive organ wasting, and rapid death in experimental models 3952.
Most critically, regarding actual human translation, a landmark 2024 study published in Circulation completely inverted the original GDF11 rejuvenation paradigm in the context of acute myocardial infarction. Utilizing a highly specific and rigorously validated liquid chromatography-tandem mass spectrometry assay, researchers proved that circulating GDF11 levels actually incline as a function of advancing age in human patients, rather than decline 40. Furthermore, in stark contrast to the original hypothesis of profound rejuvenation, boosting systemic GDF11 prior to ischemia/reperfusion injury actively exacerbated myocardial damage, accelerated pro-apoptotic signaling via the attenuation of Nkx2-5 expression, and emerged as an independent predictor of massively larger myocardial infarct sizes in humans 40. Thus, persistently high GDF11 signaling during aging is now recognized as a highly detrimental factor that compromises intrinsic cardioprotective mechanisms, highlighting the profound dangers of prematurely translating murine biological factors into human therapeutics without precise, species-specific mechanistic validation 5240.
5. Emerging Pharmacological and Genetic Interventions
As the severe limitations and toxicities of early systemic interventions become apparent, the focus of cardiovascular geroscience has shifted aggressively toward highly targeted, precision molecular approaches. The post-2023 therapeutic landscape is dominated by sophisticated metabolic modulators and precision, viral-vectored gene therapies designed to safely reverse specific hallmarks of intrinsic cardiac aging at the cellular level.
5.1 Metabolic Modulators: NAD+ Precursors
Nicotinamide adenine dinucleotide (NAD+) is an absolutely essential coenzyme for cellular energy metabolism and an indispensable obligate substrate for the sirtuin family of deacetylases (including SIRT1, SIRT2, and SIRT3) and PARP enzymes involved in genomic DNA repair. Systemic NAD+ levels precipitously decline with age across multiple tissues, fundamentally compromising mitochondrial oxidative phosphorylation and epigenetic regulation 54. Consequently, natural NAD+ precursors, specifically Nicotinamide Mononucleotide (NMN) and Nicotinamide Riboside (NR), have entered intense clinical investigation to counteract this specific metabolic decay 55.
Recent rigorous human clinical trials published in 2025 and 2026 have provided vital, long-awaited pharmacokinetic data regarding these compounds. Head-to-head, randomized comparisons reveal that daily oral supplementation with 1 gram of NMN or NR safely, sustainably, and reliably doubles circulating whole-blood NAD+ levels after 14 days, whereas generic nicotinamide provides only an acute, highly transient spike that dissipates within hours 56. Interestingly, these advanced studies indicate that orally administered NMN and NR are not primarily absorbed directly into the systemic circulation in their native, intact forms; rather, they rely heavily on complex interactions with the gut microbiome, which slowly metabolizes them into nicotinic acid - a highly potent intermediate that subsequently enters the bloodstream to elevate global NAD+ levels via the Preiss-Handler pathway 5456.
While the safety profile and tolerability of these advanced precursors are excellent, demonstrating no severe adverse events in early trials, the clinical efficacy regarding actual cardiovascular outcomes remains decidedly mixed. Short-term trials in older adults have shown some promising trends in reducing vascular stiffness and lowering diastolic blood pressure, particularly the specific MIB-626 NMN formulation 5557. However, comprehensive meta-analyses of NMN and NR trials across diverse populations have yet to demonstrate consistent, profound, disease-modifying benefits on lipid profiles, glucose control, or established structural HFpEF outcomes 55. The translational gap persists: successfully elevating whole-blood NAD+ does not guarantee adequate, sustained tissue penetration in the myocardium, nor does it ensure the functional reversal of heavily cross-linked structural myocardial senescence 5455.
5.2 Targeted Cardiac Gene Therapies: The New Frontier
The most promising, rapidly advancing frontier in treating intrinsic cardiac aging and inherited cardiomyopathies involves single-dose, targeted gene therapies delivered via highly engineered viral vectors. Rather than attempting to clumsily alter systemic biology with small molecules, these therapies utilize highly specific, cardiotropic Adeno-Associated Viruses (AAVs) to permanently correct specific molecular deficits exclusively within the cardiomyocyte.
Targeting Calcium Handling Dynamics: Given the central, undeniable role of calcium mishandling in age-related diastolic dysfunction, restoring the function of the sarcoplasmic reticulum is a primary therapeutic target. The MUSIC-HFpEF Phase 1/2a clinical trial represents a massive milestone in the field, evaluating SRD-002, an AAV1 vector explicitly designed to deliver the cardiac isoform of SERCA2a via a proprietary, minimally invasive intracoronary infusion methodology 3158. Interim data released at major cardiology congresses in 2025 demonstrated a highly favorable safety profile across cohorts receiving doses of 3E13 and 4.5E13 viral genomes, with no gene therapy-related serious adverse events at 12 months 3141. Early clinical indicators are highly promising, showing clinically meaningful stabilization and measurable improvements in pulmonary capillary wedge pressure at rest and peak exercise, alongside distinct improvements in NYHA functional class and 6-minute walk test performance, directly addressing the underlying pathophysiology of impaired myocardial relaxation 3158.
A parallel, highly innovative approach targets the specific regulatory proteins governing calcium flux. The AB-1002 program utilizes a novel, highly cardiotropic chimeric AAV2i8 capsid to deliver a constitutively active inhibitor (I-1c) of protein phosphatase 1 directly to failing cardiomyocytes 4243. By permanently inhibiting protein phosphatase 1, this genetic therapy maintains the continuous phosphorylation of phospholamban, thereby keeping endogenous SERCA2a highly active and accelerating calcium reuptake. Phase 1 trials (published in Nature Medicine) in patients with severe non-ischemic cardiomyopathy and heart failure have confirmed excellent safety, with preliminary efficacy assessments showing meaningful improvements in left ventricular ejection fraction and peak oxygen consumption, safely paving the way for the ongoing, double-blinded Phase 2 GenePHIT study, slated for completion in late 2026 25426244.
Targeting Structural Integrity and Cardiomyopathies: Beyond calcium kinetics, specific structural proteins are being targeted to reverse pathological hypertrophy and correct inherited defects that mimic advanced biological aging. The MyPeak-1 Phase 1b/2 trial is actively investigating TN-201, a first-in-class AAV-based gene therapy delivering the MYBPC3 gene to correct hypertrophic cardiomyopathy. Initial 2025 clinical data confirmed the therapy safely reaches human heart cells in vivo, measurably increases target protein levels, and safely thins the hypertrophic heart wall without triggering severe immune rejection, proving that direct genetic alteration of human myocardial structure is clinically viable 45.
Similarly, for Arrhythmogenic Cardiomyopathy - a condition causing severe structural failure and sudden death - gene replacement therapies targeting plakophilin-2 (such as RP-A601 and LX2020) and therapies restoring connexin-43 expression have shown the ability to dramatically improve electrical conductivity and restore desmosome proteins, more than doubling lifespan in preclinical models and advancing rapidly into human clinical trials 466667. Furthermore, the application of CRISPR-Cas9 genome editing has entered late-stage clinical testing. The ongoing Phase 3 MAGNITUDE trial is utilizing systemic CRISPR-Cas9 editing delivered via lipid nanoparticles to permanently silence the TTR gene in the liver, drastically reducing the production of misfolded transthyretin proteins that infiltrate and destroy heart tissue in ATTR amyloidosis with cardiomyopathy, achieving unprecedented reductions in circulating pathogenic protein levels 4769.
Table 2: Comparative Analysis of Emerging Anti-Aging Cardiovascular Interventions
| Intervention Strategy | Primary Mechanism of Action | Evidence in Animal Models | Human Clinical Evidence & Status | Current Limitations / Translational Barriers |
|---|---|---|---|---|
| Rapamycin (mTORC1 Inhibitor) | Suppresses nutrient sensing; heavily enhances macroautophagy; reduces pathogenic SASP. | Highly effective; short-term dosing yields persistent, 8-week reversals in hypertrophy and diastolic stiffness. | Extremely limited CV trials; efficacy unproven for intrinsic HFpEF reversal in humans. | Chronic use causes severe immunosuppression and insulin resistance; optimal intermittent dosing schedules completely unknown. |
| GDF11 Supplementation | TGF-beta superfamily signaling; originally purported as a universal systemic rejuvenation factor. | Highly controversial; profoundly dose-dependent. High doses universally trigger cachexia; actively exacerbates acute MI injury. | Human data conclusively shows systemic GDF11 increases with age and independently correlates with massively larger infarct sizes post-MI. | Dangerously narrow therapeutic index; immense potential for severe off-target muscular wasting and apoptotic toxicity. |
| NAD+ Precursors (NMN, NR) | Boosts systemic NAD+ reserves; enhances SIRT deacetylase activity and mitochondrial OXPHOS. | Improves metabolic flexibility; reverses endothelial senescence and vascular stiffness in aged rodents. | Readily doubles whole-blood NAD+; modest, inconsistent improvements in vascular stiffness and diastolic BP (Phase 2). | Heavily dependent on microbiome conversion to nicotinic acid; total lack of robust, long-term functional HFpEF outcomes in large human cohorts. |
| SRD-002 (SERCA2a Gene Therapy) | AAV1 vector delivery of the SERCA2a gene; directly restores diastolic active calcium reuptake in the SR. | Enhances myocardial relaxation velocity; effectively prevents age-related diastolic heart failure. | MUSIC-HFpEF trial (Phase 1/2a) confirms safety and objectively improved pulmonary capillary wedge pressures at 12 months. | Extremely high viral vector doses required for adequate myocardial transduction; immense risk of neutralizing antibodies preventing future re-dosing. |
| AB-1002 (I-1c Gene Therapy) | Chimeric AAV2i8 delivery of protein phosphatase 1 inhibitor; permanently sustains phospholamban phosphorylation. | Drastically improves cardiac function and cellular energetics in failing murine and large porcine models. | Phase 1 highly successful (safe, measurably improved LVEF); actively enrolling in the massive Phase 2 GenePHIT trial. | Long-term, multi-year durability of transgene expression remains unknown; potential for localized, destructive inflammatory responses to novel synthetic capsids. |
6. Conclusions and Future Directions
The integration of advanced geroscience with clinical cardiology has precipitated a profound and necessary paradigm shift. Intrinsic biological cardiac aging is no longer viewed as an inevitable, passive, and untreatable decline dictated by chronological time, but rather as a highly orchestrated, actively driven consequence of specific, targetable molecular mechanisms. The discovery that distinct pathways - such as PLOD3-mediated extracellular matrix hyper-cross-linking, cGAS-STING driven catastrophic inflammaging, and the SIRT2-STAT3-CDKN2B senescence axis - operate entirely independently of classical atherosclerotic risk factors demands the rapid development of distinct, highly specialized therapeutic arsenals.
Epidemiological realignments are equally critical to this effort. Data sourced from the massive INTER-CHF and ASIAN-HF registries prove conclusively that biological aging is not chronologically uniform across the globe; the severe, early-onset manifestation of fatal HFpEF in African and Southeast Asian populations highlights the urgent, non-negotiable need to look far beyond homogeneous, wealthy Western datasets and account for the diverse genetic, socioeconomic, and environmental modifiers that dictate the true pace of human aging.
While early translational attempts utilizing sweeping systemic factors like rapamycin and GDF11 encountered massive physiological barriers, biphasic toxicities, and outright clinical failures, the current generation of targeted therapies offers unprecedented, surgical precision. The ability of oral NAD+ precursors to safely leverage the microbiome to restore metabolic intermediates, coupled intimately with the early clinical success of targeted AAV gene therapies (such as SRD-002 and AB-1002) to directly and permanently reprogram cardiomyocyte calcium kinetics and structural integrity, suggests that the capacity to fundamentally slow, or even actively reverse, the human cardiac aging clock is rapidly approaching clinical reality. The ultimate, successful trajectory of this field will rely heavily on transitioning away from simplistic single-target interventions and toward highly sophisticated, combinatorial, systems-level approaches that simultaneously and safely address the deeply intertwined hallmarks of genomic instability, metabolic inflexibility, and permanent cellular senescence.