Molecular basis of major depressive disorder
Evolution of the Neurochemical Paradigm
Transition from Monoamine Deficiency to Systems Biology
For decades, the neurobiological and clinical understanding of major depressive disorder (MDD) was dominated by the monoamine hypothesis. Originally formulated in the 1960s following the serendipitous discovery of early pharmacological agents such as reserpine - which depletes monoamine stores and induces depressive symptoms - this framework posited that clinical depression was primarily caused by an absolute deficiency or chemical imbalance in synaptic serotonin (5-HT), norepinephrine, or dopamine 123. This straightforward, biologically intuitive explanation subsequently drove the development and aggressive marketing of selective serotonin reuptake inhibitors (SSRIs) throughout the 1990s, cementing the "chemical imbalance" theory in both psychiatric practice and public consciousness 23.
However, contemporary scientific consensus has decisively shifted away from this singular, reductionist framework. Comprehensive analyses of decades of literature, most notably large-scale umbrella reviews, have consistently demonstrated no empirical evidence that clinical depression is directly caused by lowered serotonin concentrations or abnormal intrinsic serotonin activity 1345. The observation that SSRIs provide therapeutic benefit to a subset of patients does not prove a primary serotonin deficit; rather, researchers now understand that acute monoamine elevation is merely an initial pharmacological trigger that initiates necessary, long-term downstream neurobiological adaptations 156. Specific lines of evidence, including studies measuring serotonin metabolites like 5-HIAA in cerebrospinal fluid and acute tryptophan depletion experiments, have repeatedly failed to reliably induce depressive symptoms in healthy controls or differentiate depressed patients from healthy cohorts 15.
Consequently, modern neuroscience classifies MDD not as a simple neurotransmitter deficit, but as a highly complex, heterogeneous, systems-level syndrome. The pathophysiology of depression arises from maladaptive neuroplastic responses to chronic stress, characterized by interconnected dysfunctions spanning structural neuroplasticity, amino acid neurotransmission, chronic neuroinflammation, neuroendocrine resistance, and altered functional connectivity within macroscopic brain circuits 3789.
Table 1 outlines the primary physiological and biochemical domains that constitute the modern integrated model of depression.
| Biological Domain | Key Molecular Targets | Pathophysiological Mechanisms in Depression |
|---|---|---|
| Neuroplasticity | BDNF, TrkB, mTORC1, MAP/ERK | Impaired synaptic remodeling; dendritic spine retraction; reduced neurogenesis in the adult hippocampus. |
| Amino Acid Neurotransmission | Glutamate, GABA, NMDA/AMPA receptors | Excitatory/inhibitory (E/I) imbalance; NMDA-mediated glutamate excitotoxicity; reduced GABAergic dendritic inhibition. |
| Neuroinflammation | IL-1β, IL-6, TNF-α, CRP, Microglia | Peripheral immune activation breaching the blood-brain barrier; M1 microglial polarization; neurotoxic cytokine release. |
| Kynurenine Metabolism | IDO, KYN, QA, 3-HK, KynA | Redirection of tryptophan from serotonin synthesis; astrocytic vs. microglial imbalance driving production of neurotoxic quinolinic acid. |
| Neuroendocrine System | Cortisol, CRH, ACTH, GR, FKBP5 | Chronic hypothalamic-pituitary-adrenal (HPA) axis hyperactivity; glucocorticoid receptor resistance; epigenetic trauma markers. |
| Microbiota-Gut-Brain Axis | SCFAs, Vagus nerve, Intestinal barrier | Dysbiosis reducing neuroprotective short-chain fatty acids; endotoxin leakage driving systemic inflammation. |
Amino Acid Neurotransmission and Synaptic Plasticity
Glutamatergic Excitotoxicity and the Quad-Partite Synapse
Recent translational research has repositioned the excitatory neurotransmitter glutamate from a peripheral consideration to a central driver in the molecular basis of depression. Magnetic resonance spectroscopy (MRS) studies have consistently demonstrated elevated levels of glutamate in specific limbic and cortical brain regions of depressed patients, including the basal ganglia and the prefrontal cortex (PFC). These elevations frequently correlate with systemic inflammatory markers and the severity of clinical anhedonia and psychomotor slowing 610.
In a healthy neural environment, the release and clearance of glutamate are tightly regulated by astrocytes, which uptake excess glutamate via specific transporters (e.g., GLT-1) to prevent toxicity 911. In depression, this clearance mechanism fails. Chronic stress induces astrocytic atrophy and downregulates glutamate transporters, leading to an accumulation of extracellular glutamate 912. High concentrations of extracellular glutamate become profoundly neurotoxic, leading to persistent overstimulation of N-methyl-D-aspartate (NMDA) receptors. This overstimulation triggers an excessive influx of intracellular calcium, generating reactive oxygen species, lipid peroxidation, and subsequent excitotoxicity that damages both neurons and supporting glia 101113.
The "connectomic glutamate framework" of depression is strongly supported by the advent of rapid-acting glutamatergic antidepressants, most notably ketamine. Unlike standard monoaminergic drugs, ketamine acts as a non-competitive antagonist at the NMDA receptor 1914. Preclinical models demonstrate that by transiently blocking NMDA receptors located primarily on GABAergic interneurons, ketamine induces a rapid disinhibition of glutamatergic pathways 1516. This process generates a controlled burst of glutamate that preferentially activates α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors 1916. The facilitation of AMPA receptors subsequently activates the mammalian target of rapamycin complex 1 (mTORC1) signaling cascade, driving the rapid synthesis of synaptic proteins and the immediate release of Brain-Derived Neurotrophic Factor (BDNF) 916. This molecular cascade culminates in the rapid genesis of new dendritic spines and the restoration of synaptic connectivity in the prefrontal cortex within hours, offering profound clinical relief for treatment-resistant depression (TRD) 916.
GABAergic Deficits and Neurosteroid Modulation
Running parallel to glutamatergic hyperactivity is a pronounced deficit in the central nervous system's primary inhibitory neurotransmitter, γ-aminobutyric acid (GABA). Magnetic resonance spectroscopy and postmortem tissue analyses reveal significant reductions in cortical GABA concentrations and a specific depletion in the density of GABAergic interneurons - particularly somatostatin (SST) and parvalbumin (PV) expressing cells - in the prefrontal cortex following chronic stress exposure 181517. This reduction compromises dendritic inhibition and directly contributes to the overarching excitatory/inhibitory (E/I) imbalance that characterizes depressive neural circuitry 1518.
The pathophysiological relevance of GABAergic signaling has been further elucidated by exploring specific GABAA receptor subunits, such as α5-GABAARs and δ-GABAARs 1419. The therapeutic potential of targeting these receptors was validated by the development of brexanolone, the first FDA-approved treatment specifically for peripartum depression 141819. Brexanolone is a synthetic formulation of allopregnanolone, an endogenous neurosteroid that functions as a positive allosteric modulator (PAM) of GABAA receptors. Chronic stress heavily depletes intrinsic neurosteroid signaling, impairing the brain's inhibitory tone; the administration of neurosteroid-based treatments restores GABAA signaling, dampens hyperactive neural circuits, and rapidly alleviates both affective and cognitive depressive symptoms 1141820. Curiously, preclinical data indicate that both positive allosteric modulators (PAMs, such as GL-II-73) and negative allosteric modulators (NAMs) targeting α5-GABAARs can exhibit antidepressant properties, suggesting that both the enhancement of dendritic inhibition and the acute, transient resetting of receptor activity can facilitate recovery from stressed states 141819.
Brain-Derived Neurotrophic Factor and TrkB Signaling
The structural degradation of synapses caused by E/I imbalance is inextricably linked to deficits in neurotrophic support. Brain-Derived Neurotrophic Factor (BDNF) is the most abundant neurotrophin in the adult brain, responsible for inducing neurogenesis, supporting neuronal survival, and modulating long-term potentiation (LTP) 212223. In healthy physiology, mature BDNF binds with high affinity to the tropomyosin receptor kinase B (TrkB). This activation triggers vital intracellular signaling cascades, including the phosphatidylinositol 3-kinase (PI3K)/Akt pathway and the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway, which govern the structural plasticity of the central nervous system 721.
Extensive meta-analyses confirm that serum and cerebral BDNF levels are significantly attenuated in depressed patients, directly correlating with the severity of clinical symptoms and observed reductions in hippocampal volume 1317222324. Elevated glucocorticoids, resulting from chronic stress, directly suppress the transcription of the BDNF gene 1725. Furthermore, depression alters the delicate processing balance between precursor proBDNF and mature BDNF. While mature BDNF supports neuroprotection via TrkB, proBDNF binds to the p75 neurotrophin receptor (p75NTR), facilitating long-term depression (LTD) and initiating apoptotic programmed cell death 23. Elevated proBDNF and diminished mature BDNF drive the relentless synaptic pruning and dendritic retraction seen in the depressed brain 2326. Conventional antidepressant pharmacotherapies incrementally reverse these deficits over weeks, upregulating BDNF expression and slowly reconstructing eroded synaptic architectures 131724.
Neuroinflammation and Immune-Brain Interactions
Cytokine Dynamics and Blood-Brain Barrier Permeability
Over the past two decades, extensive clinical and epidemiological evidence has established neuroinflammation as a critical transdiagnostic driver of depression and treatment resistance. Individuals diagnosed with MDD consistently exhibit elevated peripheral blood concentrations of acute-phase proteins and pro-inflammatory cytokines, most robustly C-reactive protein (CRP), Interleukin-6 (IL-6), Interleukin-1 beta (IL-1β), and Tumor Necrosis Factor-alpha (TNF-α) 81725272728. High systemic inflammation often predicts a poorer response to conventional monoaminergic antidepressants, necessitating targeted or precision interventions 1029.
Under conditions of chronic psychosocial or physiological stress, the integrity of the blood-brain barrier (BBB) becomes compromised, allowing peripheral inflammatory signals and immune cells to infiltrate the central nervous system 83031. Upon infiltration, these signals interact with microglia, the resident macrophages of the brain. Microglia respond to damage-associated molecular patterns (DAMPs) and stress molecules by shifting from a ramified, surveying state into a pro-inflammatory, "M1-like" active phenotype 7928. Positive emission tomography (PET) imaging utilizing translocator protein (TSPO) ligands confirms widespread microglial activation in the anterior cingulate cortex and temporal regions of patients with MDD 916. Once activated, microglia secrete further cytokines, propagating local neuroinflammation that disrupts monoamine synthesis, directly impairs BDNF production, and attacks synaptic structures 9252829.
The Tryptophan-Kynurenine Metabolic Shift
The most clearly delineated molecular pathway linking systemic immune activation to depressive neurotoxicity is the disruption of the kynurenine (KYN) pathway of tryptophan metabolism. Under homeostatic conditions, the essential amino acid tryptophan serves as the primary biochemical precursor for the synthesis of central serotonin 3233. However, the presence of pro-inflammatory cytokines (such as IL-1β, IL-6, and particularly interferon-gamma) robustly induces the activation of the enzyme indoleamine 2,3-dioxygenase (IDO) in peripheral tissues, endothelial cells, and central glial cells 7253233.
IDO activation aggressively shunts tryptophan away from serotonin synthesis and redirects it down the kynurenine metabolic cascade. Consequently, blood and cerebrospinal fluid ratios of kynurenine to tryptophan increase significantly in depressed populations, serving as a direct biomarker of IDO activity and systemic inflammation 323334. Circulating kynurenine readily permeates the blood-brain barrier, where it is metabolized along two divergent and highly competitive branches governed by local glial cell dynamics 113032:
- The Neurotoxic Microglial Branch: In active, pro-inflammatory microglia, the enzyme kynurenine monooxygenase (KMO) converts kynurenine into 3-hydroxykynurenine (3-HK), which is subsequently degraded into quinolinic acid (QA). QA is a potent, direct endogenous agonist of the NMDA receptor. Its accumulation causes massive glutamatergic excitotoxicity, generates severe oxidative stress via lipid peroxidation, and ultimately drives neuronal apoptosis and the structural atrophy associated with depression 1125303234.
- The Neuroprotective Astrocytic Branch: In resting astrocytes, the enzyme kynurenine aminotransferase (KAT) converts kynurenine into kynurenic acid (KynA). KynA operates as a competitive antagonist at the NMDA receptor, effectively mitigating the effects of glutamate spillover and providing vital neuroprotection 113234.
During major depressive episodes, the overarching inflammatory state heavily skews this critical balance. Dysfunctions in regulatory enzymes, such as aminocarboxymuconate-semialdehyde decarboxylase (ACMSD), fail to divert intermediates toward protective endpoints 30. The KynA/QA ratio plummets, resulting in a synaptic microenvironment flooded with neurotoxic quinolinic acid. The integrated pathology of depression can therefore be conceptualized as a continuous loop: chronic stress induces hypercortisolism and systemic inflammation, leading to microglial activation. Pro-inflammatory cytokines trigger the indoleamine 2,3-dioxygenase (IDO) enzyme, shifting tryptophan metabolism toward the kynurenine pathway. This yields quinolinic acid, provoking glutamatergic excitotoxicity via NMDA receptors, while parallel reductions in BDNF impair TrkB signaling and synaptic plasticity 79112135.
Neuroendocrine Dysregulation and Epigenetic Mechanisms
Hypothalamic-Pituitary-Adrenal Axis Hyperactivity
The biological response to stress is governed by the hypothalamic-pituitary-adrenal (HPA) axis, a highly calibrated neuroendocrine system that is profoundly dysregulated in 50% to 80% of individuals suffering from major depression 353637. In response to acute psychological or physiological stressors, the hypothalamus secretes corticotropin-releasing hormone (CRH). CRH stimulates the anterior pituitary gland to release adrenocorticotropic hormone (ACTH), which travels through the bloodstream to prompt the adrenal cortex to synthesize and secrete glucocorticoids, primarily cortisol in humans 2738.
Under normal physiological parameters, elevated cortisol binds to glucocorticoid receptors (GR) concentrated in the hippocampus, hypothalamus, and pituitary. This binding triggers a crucial ultra-short negative feedback loop, inhibiting further secretion of CRH and ACTH, and effectively terminating the stress response 2730353940. In MDD, this vital feedback mechanism is functionally shattered. The resultant continuous HPA axis hyperactivity subjects the brain to chronic hypercortisolism. Sustained exposure to high levels of cortisol acts as a profound neurotoxin; it drastically downregulates BDNF expression, halts the proliferation of neural stem cells in the dentate gyrus, and induces the severe structural atrophy of the hippocampus observed in chronic, treatment-resistant depression 17253038.
Glucocorticoid Receptor Resistance and FKBP5 Methylation
The prevailing molecular explanation for unyielding HPA axis hyperactivity is the "glucocorticoid resistance model." Chronic stress and continuous cortisol exposure induce a functional impairment, downregulation, or desensitization of the glucocorticoid receptor (GR). This resistance renders the HPA axis deaf to the suppressive signals of its own circulating hormones, allowing cortisol levels to remain perpetually elevated 2730353640.
Crucially, glucocorticoid resistance establishes a perilous bidirectional interplay with the immune system. Ordinarily, glucocorticoids exert potent anti-inflammatory effects by suppressing cytokine synthesis. However, when systemic tissues develop glucocorticoid resistance, inflammatory pathways escape normal endocrine control, allowing pro-inflammatory cytokines to proliferate unabated 27353640. Elevated cytokines, particularly IL-6 and TNF-α, further inhibit the translocation of the GR complex into the cell nucleus, exacerbating the resistance and locking the organism into a self-perpetuating cycle of neuroinflammation and hypercortisolism 3035.
This resistance is heavily mediated by epigenetic factors and gene-environment interactions. A primary focal point of this research is the FKBP5 gene, which encodes the FK506-binding protein 51, a co-chaperone of heat shock protein 90 (HSP90). FKBP5 binds to the glucocorticoid receptor complex and actively reduces its affinity for cortisol and delays its translocation into the nucleus, acting as a major negative regulator of GR sensitivity 30364142. Exposure to childhood maltreatment, trauma, or severe early-life stress induces long-lasting epigenetic alterations, specifically the demethylation (hypomethylation) of regulatory regions within the FKBP5 gene 3639424344. This trauma-induced hypomethylation leads to the over-expression of FKBP5 protein, significantly reducing GR sensitivity and effectively hardwiring glucocorticoid resistance into the patient's physiology 394244. Similarly, hypermethylation of the NR3C1 gene, which encodes the glucocorticoid receptor itself, has been identified in individuals with major depression and a history of childhood trauma, further compounding the dysfunction of the stress response system across the lifespan 414243.
Genomic Architecture and Population Diversity
Multi-Ancestry Genome-Wide Association Studies
The genomic architecture of major depressive disorder is extraordinarily complex and highly polygenic. MDD does not stem from a single, highly penetrant vulnerability gene; rather, the genetic risk is distributed across hundreds or thousands of single nucleotide polymorphisms (SNPs), each exerting a microscopic effect on the overall liability 3845.
Historically, Genome-Wide Association Studies (GWAS) targeting psychiatric disorders have been severely limited by a near-exclusive reliance on cohorts of European descent. This methodological bottleneck raised significant concerns regarding the biological completeness of the findings and their clinical transferability to global populations, risking the exacerbation of health inequalities 45464847. Addressing this critical gap, a landmark multi-ancestry GWAS was published in 2024 and 2025, utilizing aggregated genomic data from over five million individuals across 29 countries. The primary analytical cohort comprised 88,316 MDD cases and 902,757 controls, and crucially featured unprecedented diversity: 36% of the effective sample size was of African ancestry, 32% Hispanic/Latin American, 26% East Asian, and 6% South Asian 464848.
This expansive, multi-ancestry analysis successfully identified 53 novel genetic risk loci significantly associated with depression, alongside a transcriptome-wide association study that prioritized 205 novel risk genes 464848. The identified genetic variants mapped heavily to regions governing neuronal development, synaptic transmission, and emotional regulation circuitry, reinforcing the conceptualization of depression as a structural disease of the brain 4547. Most importantly, the research team developed a metric called power-adjusted transferability (PAT) to evaluate the predictive power of genetic risk scores across ethnicities. The analysis revealed that polygenic risk profiles developed exclusively from European cohorts possessed surprisingly low transferability to other populations: only 27% transferability to samples of African ancestry, 29% to East Asian ancestry, and 63% to Hispanic/Latin American ancestry 4848. These findings underscore that maximizing ancestral diversity is an absolute prerequisite for unraveling the core, universal genetic drivers of depression.
Gene-Environment Interactions
Genetic vulnerability to depression is not deterministic but is strongly mediated through gene-environment (GxE) interactions 49. The most thoroughly studied example involves polymorphisms in the promoter region of the serotonin transporter gene (SLC6A4, specifically the 5-HTTLPR polymorphism). Meta-analyses indicate that individuals carrying the short (S) allele exhibit a significantly heightened risk of developing depression, but this risk is primarily actualized only when the individual is exposed to severe environmental stressors, such as childhood maltreatment or chronic medical conditions 2749. While some analyses report inconsistencies regarding this specific interaction depending on the self-reporting methodology used for stress exposure, the overarching consensus remains that underlying genetic susceptibility across monoaminergic, neurotrophic, and HPA axis genes must intersect with environmental epigenetic modulators to produce clinical depression 2749.
The Microbiota-Gut-Brain Axis
Short-Chain Fatty Acids and Neural Homeostasis
A rapidly expanding frontier in understanding the molecular pathology of depression centers on the Microbiota-Gut-Brain (MGB) axis. MDD is consistently associated with pronounced intestinal dysbiosis, characterized by a marked reduction in microbial biodiversity, an increase in opportunistic pathogenic bacteria, and increased intestinal permeability 28315051.
The primary molecular mediators of communication across the MGB axis are short-chain fatty acids (SCFAs), primarily acetate, propionate, and butyrate. These metabolites are generated in the large intestine through the anaerobic bacterial fermentation of indigestible dietary fibers and resistant starches 505253. SCFAs possess potent neuroactive and immunomodulatory properties. Upon production, they enter the systemic circulation and traverse the blood-brain barrier (BBB) via specific monocarboxylate transporters 315253.
At physiological concentrations, SCFAs maintain the integrity of the BBB by upregulating tight junction proteins, protecting the central nervous system from peripheral toxins 5256. Within the brain, SCFAs act directly upon the neural immune landscape. By binding to G protein-coupled receptors (GPCRs) such as free fatty acid receptors 2 and 3 (FFAR2 and FFAR3), and by functioning as potent inhibitors of histone deacetylases (HDACs), SCFAs suppress microglial activation and regulate the recruitment of T cells and macrophages, thereby exerting profound anti-inflammatory effects 505356. Preclinical studies employing high-fiber diets demonstrate that elevated serum SCFA levels (particularly acetate) are inversely correlated with hippocampal neuroinflammation, significantly reducing levels of IL-1β, IL-6, and TNF-α. Concurrently, acetate acts as a strong mediator for increased BDNF production, enhancing adult hippocampal neurogenesis and reversing depressive-like behaviors 315052.
Conversely, the gut dysbiosis observed in depression severely diminishes the endogenous production of SCFAs. This reduction compromises the intestinal epithelial barrier ("leaky gut"), permitting bacterial endotoxins such as lipopolysaccharides (LPS) to enter the bloodstream. This endotoxemia triggers the systemic inflammatory cascades that drive the cytokine-mediated tryptophan-kynurenine shifts discussed earlier 28345051. Alongside this metabolic pathway, the MGB axis communicates rapidly with the brain via the vagus nerve. The cholinergic anti-inflammatory pathway (CAIP) of the vagus nerve normally suppresses peripheral immune responses; however, chronic stress disrupts this neural-immune interaction, stripping the body of another vital regulatory mechanism and further aggravating treatment-resistant depressive states 3053. The interplay between metabolic dysregulation, insulin resistance (frequently measured via the TyG-BMI index), and systemic inflammation further highlights that depression is a whole-body metabolic syndrome as much as a localized brain disorder 5455.
Network-Level Dysfunction and Neuroimaging Biotypes
Large-Scale Brain Networks
The microscopic molecular deficits detailed above - synaptic pruning driven by BDNF depletion, glutamatergic excitotoxicity, and microglial inflammation - do not manifest in isolation. Instead, they compound to degrade the structural and functional integrity of large-scale macroscopic brain networks. The clinical presentation of depression is ultimately governed by the destabilization of functional interactions among the brain's core networks: the Default Mode Network (DMN, heavily active during introspection and rumination), the Salience Network (responsible for threat detection and emotional processing), and the Central Executive/Cognitive Control Network (governing attention, working memory, and goal-directed behavior) 95657.
Because depression encompasses a staggering array of genetic vulnerabilities and environmental triggers, the resulting network dysfunctions are highly heterogeneous. Different patients exhibit distinctly different patterns of functional dysconnectivity, which explains the vast diversity in symptom profiles and the notoriously unpredictable, trial-and-error response rates to conventional monoaminergic antidepressants 58596061.
Functional Magnetic Resonance Imaging Biotypes
In 2024, a landmark multi-cohort investigation published in Nature Medicine successfully bridged the gap between neurobiology and personalized clinical care. By utilizing functional magnetic resonance imaging (fMRI) combined with sophisticated machine learning cluster analysis, researchers assessed task-free and task-evoked brain activity in 801 participants diagnosed with clinical depression and anxiety 606162. The study precisely quantified functional connectivity and regional activation within six previously established networks: the default mode circuit, salience circuit, attention circuit, negative affect circuit, positive affect circuit, and cognitive control circuit 596063.
This data-driven approach bypassed traditional subjective diagnostic boundaries, yielding six clinically distinct, transdiagnostic biological subtypes, or "biotypes," of depression and anxiety 586764. Crucially, during a subset trial of 250 randomized participants, these biotypes strongly and accurately predicted differential responses to standard pharmacological interventions (escitalopram, sertraline, or venlafaxine) and behavioral problem-solving therapy 58596269.
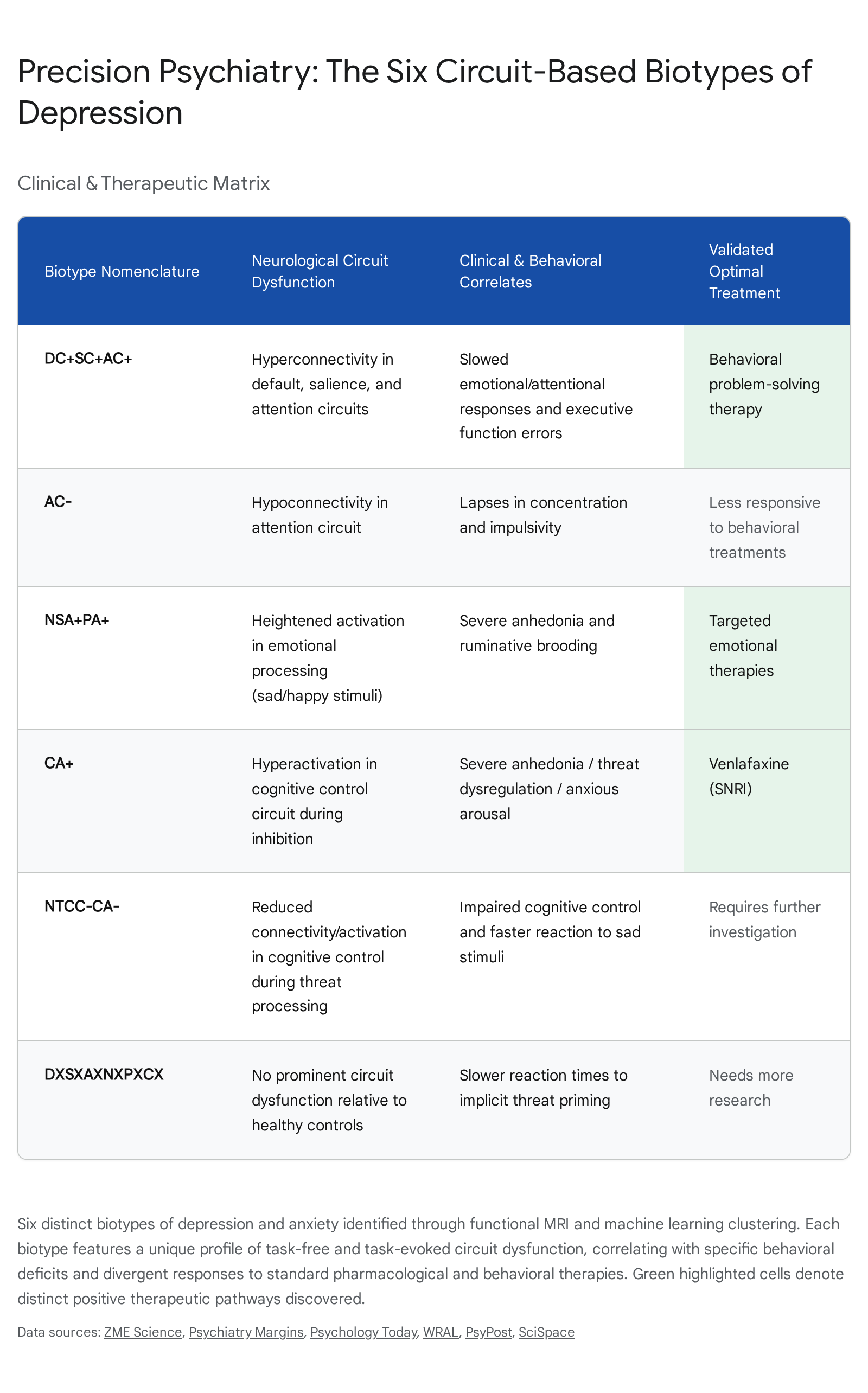
The specific profiles and therapeutic implications of these six fMRI biotypes are detailed as follows:
- Biotype DC+SC+AC+: Characterized by significant baseline hyperconnectivity across the default mode, salience, and attention circuits while at rest. Clinically, these patients exhibited slowed behavioral responses to target stimuli and sad faces, yet demonstrated relatively few errors in cognitive control tasks. Notably, this hyper-connected biotype responded most favorably to behavioral problem-solving therapy, likely because their active circuitry was primed to engage with and integrate new cognitive skills 6367646566.
- Biotype CA+: Defined by pronounced hyperactivation specifically within the cognitive control circuit during task-evoked inhibition. Individuals within this cluster suffered from severe clinical anhedonia, elevated anxious arousal, negative bias, and dysregulation in threat response, corresponding with high error rates in sustained attention tasks. This specific biotype exhibited the best clinical response to the serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine 62636466.
- Biotype AC - : Marked by distinct hypoconnectivity isolated to the frontoparietal attention circuit. Behaviorally, these patients struggled with prominent lapses in concentration and impulsivity, making more errors on cognitive measures despite faster response times. Unlike the hyperconnected DC+SC+AC+ group, this biotype was significantly less responsive to behavioral talk therapy interventions 59696566.
- Biotype NSA+PA+: Characterized by heightened regional activation within emotional processing centers - specifically, hyperactivation of the negative affect circuit when exposed to sad stimuli, and hyperactivation of the positive affect circuit when exposed to happy stimuli. Clinically, these individuals experienced severe, prominent anhedonia and ruminative brooding, necessitating more targeted emotional regulation therapies 5962656667.
- Biotype NTCC - CA - : A smaller, highly specific cluster differentiated by a loss of functional connectivity within negative emotion circuits during the conscious processing of threatening stimuli, alongside reduced activation in cognitive control circuits. Behaviorally, they exhibited impaired cognitive control but notably faster reaction times to sad stimuli, alongside lower levels of ruminative brooding. Optimal treatment pathways for this biotype require further targeted investigation 596668.
- Biotype DXSXAXNXPXCX: Intriguingly, this biotype demonstrated completely intact activation and connectivity, showing no prominent or statistically significant circuit dysfunction in the regions imaged relative to healthy controls. These patients only exhibited slightly slower reaction times to implicit threat priming. This finding suggests that the specific biological driver for this subset of patients involves mechanisms (such as highly localized neuroinflammation or purely metabolic variables) that current macroscopic fMRI parameters do not capture 59666869.
By quantifying depression through objective measures of brain circuit dysfunction, this biotype framework represents a foundational shift in clinical management. It provides a robust, evidence-based mechanism to move psychiatry away from the antiquated, trial-and-error prescription of monoamine modulators and toward the realization of precision medicine - where interventions are rationally matched directly to the underlying molecular, cellular, and connectomic deficits of the individual patient 606162.
Conclusion
The scientific understanding of the molecular basis of major depressive disorder has advanced decisively beyond the simplistic paradigm of monoamine deficiency. Today, MDD is recognized as a profound, multifactorial, and highly heterogeneous systemic disorder of the brain. Its pathogenesis is deeply rooted in the continuous erosion of structural neuroplasticity - driven by the depletion of Brain-Derived Neurotrophic Factor and severely impaired TrkB signaling - and is exacerbated by a destructive imbalance between glutamatergic excitotoxicity and GABAergic dendritic inhibition. These synaptic failures are heavily orchestrated by the brain's surrounding immune environment, where chronic psychosocial stress induces HPA axis hypercortisolism, glucocorticoid receptor resistance, and aggressive systemic inflammation. Pro-inflammatory cytokines ultimately hijack peripheral and central tryptophan metabolism, generating neurotoxic kynurenine metabolites that actively degrade neural networks from within.
This pathological cascade is underpinned by a massive, diverse polygenic architecture and critical epigenetic vulnerabilities formed by early-life trauma, while being continuously modulated by the systemic metabolic output of the gut microbiome. Ultimately, these microscopic disruptions manifest as measurable macroscopic dysfunctions in large-scale brain networks. The recent identification of objective fMRI biotypes conclusively proves that depression is not a single clinical entity, but a collection of distinct neurobiological phenotypes. Moving forward, the integration of multi-omics data, inflammatory biomarkers, and advanced neuroimaging promises to transform the treatment of depression into a rigorous discipline of precision medicine, targeting the precise molecular and circuit-level deficits unique to each individual patient.