Mitophagy in Aging and Neurodegeneration
Introduction to Mitochondrial Homeostasis
Mitochondria are the principal organelles responsible for eukaryotic cellular bioenergetics, generating the vast majority of cellular adenosine triphosphate (ATP) through the process of oxidative phosphorylation. Beyond their role as cellular powerhouses, mitochondria act as central signaling hubs that regulate lipid and amino acid metabolism, calcium homeostasis, and intrinsic apoptotic pathways 12. Evolutionary biology dictates that these organelles originated from endosymbiotic proteobacteria, a heritage that explains their double-membrane structure and the presence of their own circular genome, mitochondrial DNA (mtDNA) 12.
Because the electron transport chain inherently leaks electrons, mitochondria are the primary intracellular source of reactive oxygen species (ROS). Under normal physiological conditions, these free radicals are neutralized by robust endogenous antioxidant defense mechanisms. However, cumulative exposure to oxidative stress, environmental toxins, and genomic mutations can damage mitochondrial structures 23. Over time, this results in a population of dysfunctional mitochondria characterized by depolarized membranes, inefficient ATP generation, excessive ROS production, and the pathological leakage of mtDNA into the cytosol 24.
To avert the catastrophic consequences of bioenergetic failure and oxidative toxicity, eukaryotic cells possess an elaborate mitochondrial quality control system. Mitophagy - a highly conserved, selective form of macroautophagy - is the primary autolysosomal clearance mechanism by which cells sequester and degrade damaged, superfluous, or aged mitochondria 56. The precision of this quality control mechanism is exceptionally critical in post-mitotic cells with extreme metabolic demands, such as neurons and skeletal myocytes, which lack the capacity to simply dilute damaged organelles through cellular division 78. Defective mitophagy is now recognized as a central driver of human pathology, inextricably linked to age-related neurodegenerative diseases (including Parkinson's disease and Alzheimer's disease), metabolic syndromes, and the broader physiological decline associated with biological aging 379. Consequently, the molecular architecture of mitophagy and its pharmacological modulation have emerged as paramount focuses in geromedicine.
Molecular Machinery of Macroautophagy and Mitophagy
The execution of mitophagy relies heavily on the core molecular machinery of macroautophagy, functioning in concert with specific receptor-mediated or ubiquitin-dependent signals that confer cargo selectivity.
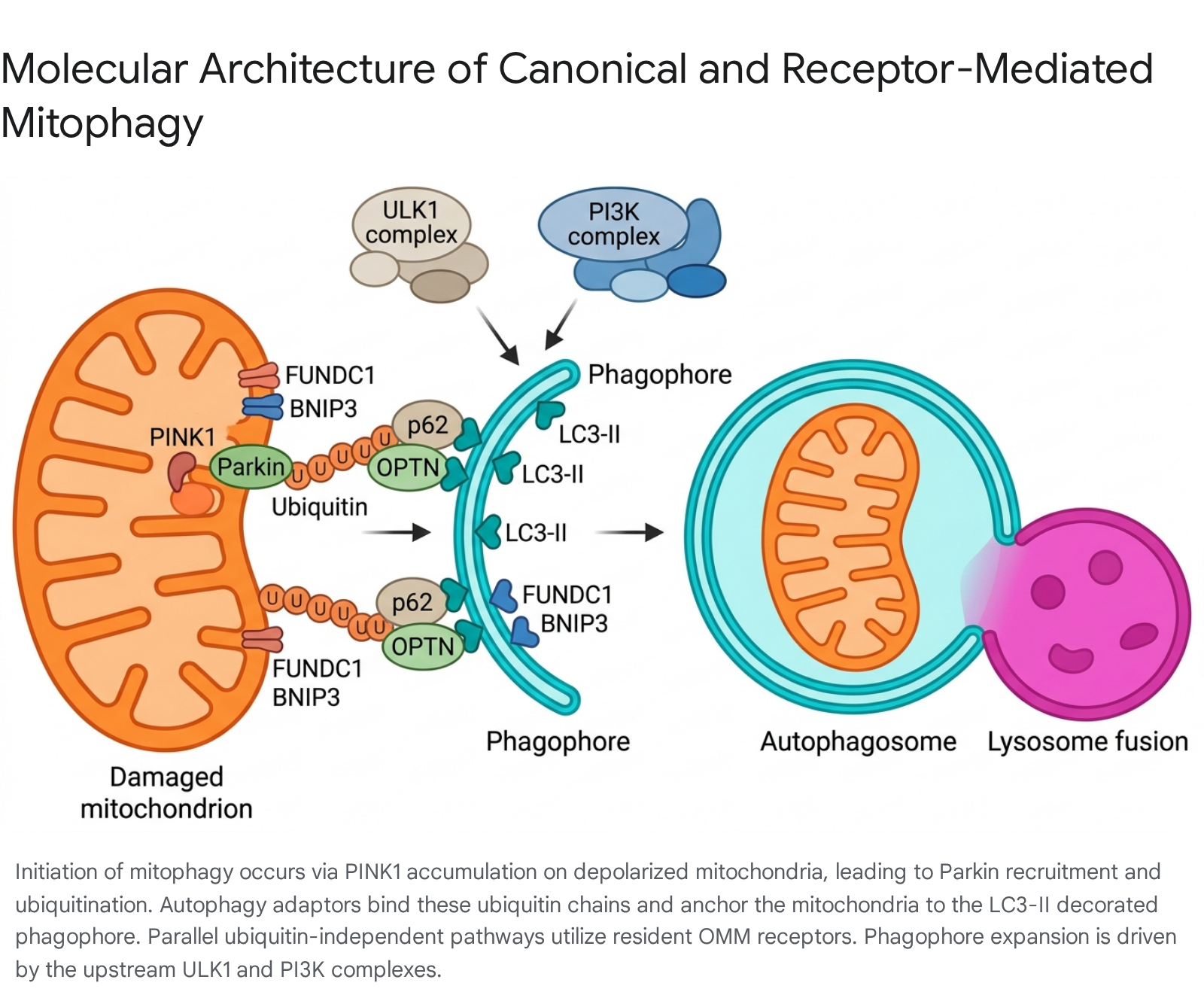
The autophagic process is traditionally divided into four distinct phases: initiation, nucleation, expansion and maturation, and lysosomal fusion 1011.
Core Autophagy Protein Complexes
The initiation of the autophagic cascade is tightly coupled to the cell's nutrient and energy status, governed primarily by the AMP-activated protein kinase (AMPK) and the mammalian target of rapamycin complex 1 (mTORC1) 1112. During periods of energy stress or hypoxia, AMPK is activated and subsequently phosphorylates and activates the Unc-51-like kinase 1 (ULK1) complex. This initiation complex consists of the serine/threonine kinases ULK1/2, the regulatory subunit ATG13, the scaffold protein FIP200 (also known as RB1CC1), and ATG101 101113. Conversely, in nutrient-rich states, mTORC1 hyperphosphorylates ULK1 and ATG13, effectively suppressing autophagosome formation 101113.
Once initiated, the nucleation of the phagophore (the initial isolation membrane) is driven by the class III phosphatidylinositol 3-kinase (PI3K) complex. This crucial complex comprises VPS34 (the lipid kinase), VPS15, Beclin-1 (VPS30/ATG6), and ATG14 101114. The PI3K complex localized to the phagophore assembly site generates phosphatidylinositol-3-phosphate (PI3P). The local accumulation of PI3P acts as a biochemical beacon, recruiting downstream effector proteins, such as WIPI2, that are essential for membrane elongation 101314.
Phagophore expansion and structural maturation are coordinated by two highly conserved ubiquitin-like conjugation systems. The first system utilizes the E1-like enzyme ATG7 and the E2-like enzyme ATG10 to covalently conjugate the ubiquitin-like protein ATG12 to ATG5. The ATG12 - ATG5 conjugate then binds to ATG16L1, forming a massive multimeric complex (ATG12 - ATG5 - ATG16L1) 1013. This trimeric complex functions as an E3-like ligase for the second conjugation system, which facilitates the lipidation of the cytosolic protein LC3 (or other ATG8-family proteins) 101114. LC3 is cleaved by the protease ATG4 and then conjugated to the lipid phosphatidylethanolamine (PE) via the actions of ATG7 and ATG3, forming LC3-II 1014. LC3-II is incorporated into both the inner and outer leaflets of the expanding phagophore, where it plays a critical structural role in sealing the double-membrane vesicle and acts as a docking site for selective cargo receptors 1011.
The vast amounts of lipid membrane required to envelop a mitochondrion are supplied via ATG9-carrying transport vesicles and ATG2-associated lipid transfer channels 1113. Following complete encapsulation of the mitochondrion, the mature autophagosome must fuse with a late endosome or lysosome. This final degradative step is mediated by specific SNARE complex proteins, including syntaxin 17 (STX17), YKT6, and RAB7, allowing the acidic hydrolases of the lysosome to break down the mitochondrial cargo 1011.
| Functional Stage | Associated Core Proteins | Primary Biochemical Function |
|---|---|---|
| Initiation | ULK1/2, ATG13, FIP200, ATG101 | Senses metabolic stress via AMPK/mTORC1; triggers phagophore assembly. |
| Nucleation | VPS34, VPS15, Beclin-1, ATG14 | Generates PI3P at the phagophore assembly site to recruit lipid-binding effectors. |
| Expansion | ATG12 - ATG5 - ATG16L1, ATG7, ATG3 | Acts as an E3-like ligase system driving the conjugation of LC3 to PE. |
| Maturation | LC3 (ATG8 family), ATG9, ATG2 | LC3-II anchors cargo receptors; ATG2/ATG9 mediate lipid transfer for membrane elongation. |
| Fusion | STX17, YKT6, RAB7 | Facilitates the tethering and fusion of the autophagosome with the lysosome. |
The Canonical PINK1-Parkin Pathway
While the core macroautophagy machinery constructs the degradative vesicle, cargo selectivity is achieved through specific targeting mechanisms. The most thoroughly characterized pathway for damage-induced mitophagy relies on PTEN-induced putative kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin (PRKN) 915. Under physiological baseline conditions, PINK1 is continuously imported through the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM), where it undergoes constitutive cleavage by presenilins-associated rhomboid-like (PARL) proteases, followed by rapid proteasomal degradation via the N-end rule pathway 15.
However, when a mitochondrion suffers stress that dissipates its transmembrane potential, the import of PINK1 is arrested. Consequently, full-length PINK1 accumulates on the OMM, where it undergoes homodimerization and autophosphorylation, leading to its functional activation 1516. Active PINK1 subsequently phosphorylates pre-existing, basal ubiquitin molecules on the OMM at Serine 65. These phosphorylated ubiquitin chains serve as highly specific recruitment signals for cytosolic Parkin, which exists in an auto-inhibited state 1516. Once recruited, Parkin is also directly phosphorylated by PINK1, which relieves its auto-inhibition and fully unleashes its E3 ligase activity 1516.
Activated Parkin catalyzes the extensive polyubiquitination of numerous OMM-resident proteins, including voltage-dependent anion channel 1 (VDAC1) and mitofusins 716. This massive ubiquitin coat acts as an amplification loop, attracting specialized selective autophagy adaptor proteins - most notably Optineurin (OPTN), Nuclear Domain 10 Protein 52 (NDP52), and p62 (SQSTM1) 91517. These adaptors are defined by the presence of two crucial domains: a ubiquitin-binding domain that attaches to the Parkin-ubiquitinated mitochondrion, and an LC3-interacting region (LIR) that docks with LC3-II on the inner surface of the expanding phagophore 1516. Through this dual binding capability, the adaptors physically tether the damaged mitochondrion to the autophagic membrane, ensuring precise encapsulation.
Receptor-Mediated Mitophagy Pathways
In parallel to the ubiquitin-dependent PINK1/Parkin system, cells utilize ubiquitin-independent mechanisms mediated by specific mitophagy receptors localized to the mitochondrial membranes. These receptors possess intrinsic LIR motifs, allowing them to bind directly to LC3-II and bypass the need for extensive ubiquitination by Parkin 715.
Key OMM receptors include BNIP3, NIX (BNIP3L), and FUNDC1. These receptors are heavily involved in programmed, physiological mitophagy rather than purely damage-response mitophagy. For instance, NIX is absolutely essential for the developmentally programmed elimination of mitochondria during the terminal differentiation of reticulocytes into mature erythrocytes 15. FUNDC1 is primarily regulated by hypoxia; under normoxic conditions, it is phosphorylated and inactive, but severe hypoxia triggers its dephosphorylation by PGAM5, vastly increasing its affinity for LC3-II and inducing rapid mitochondrial clearance 615. Other mammalian OMM receptors identified in recent literature include BCL2L13, FKBP8, and AMBRA1 9.
Furthermore, inner mitochondrial membrane (IMM) proteins can also function as mitophagy receptors. Prohibitin 2 (PHB2) and the phospholipid cardiolipin normally reside within the IMM. However, upon severe mitochondrial damage that results in the rupture of the OMM, these IMM components are exposed to the cytosol, where they directly bind LC3 to trigger the engulfment of the compromised organelle 15.
Non-Canonical and ATG5-Independent Pathways
Emerging research indicates that the mitophagic machinery possesses remarkable plasticity and redundancy, capable of operating even when core lipidation components are disrupted. Investigations utilizing nanobody-based mitochondrial targeting systems (such as the mito-QC assay) have demonstrated that while the forced mitochondrial recruitment of ULK1 or LC3 variants induces a conventional, ATG5-dependent mitophagy pathway, alternative routes exist 518.
Specifically, targeted recruitment of an ATG16L1-derived peptide (which retains binding affinity for FIP200 and WIPI2) successfully induces an unconventional form of mitophagy. This alternative pathway entirely bypasses the absolute requirement for ATG5 and the subsequent lipidation of LC3 518. While still dependent on the upstream activity of FIP200 and the PI3K/VPS34 complex, this ATG5-independent route does not form a canonical autophagosome. Instead, it delivers targeted mitochondria directly to early endosomes, which subsequently traffic to and fuse with lysosomes 518. The existence of such bypass mechanisms highlights the evolutionary imperative of mitochondrial clearance, ensuring that cells maintain alternative fail-safes when canonical autophagy is chemically or genetically compromised.
Mitophagy Dysfunction in Neurodegenerative Diseases
Neurons face unique morphological and bioenergetic challenges. Because they are post-mitotic and highly polarized - often featuring axonal extensions that span vast distances - they rely almost entirely on oxidative phosphorylation rather than glycolysis for survival 317. Maintaining healthy synapses requires local mitochondrial quality control and continuous, bidirectional axonal transport of these organelles. When mitophagy stalls, neurons accumulate damaged mitochondria that fail to supply ATP and release oxidative radicals, directly lowering the apoptotic threshold and triggering programmed cell death 317.
Parkinson's and Alzheimer's Diseases
The pathogenesis of Parkinson's disease (PD) provided the foundational link between neurodegeneration and mitophagy. Loss-of-function mutations in the PINK1 and PRKN (Parkin) genes are established drivers of early-onset, autosomal recessive forms of PD 915. Without the functional PINK1/Parkin axis, dopaminergic neurons in the substantia nigra cannot clear oxidatively damaged mitochondria, leading to profound energy crises, cellular death, and the hallmark aggregation of alpha-synuclein into Lewy bodies 19. Research further demonstrates that the failure of autophagic pathways leads to hyperactivation of NAD+-consuming enzymes, depleting intracellular NAD+ pools and independently triggering mitochondrial depolarization 21.
In Alzheimer's disease (AD), defective mitophagy correlates tightly with the accumulation of both amyloid-beta (Aβ) plaques and hyperphosphorylated tau tangles 3717. As the brain ages, cumulative ROS production directly oxidizes core mitophagy proteins, including Parkin and LC3, structurally impairing their ability to execute mitochondrial clearance 3. Furthermore, AD pathology often disrupts downstream lysosomal processes. Familial AD mutations in the presenilin 1 (PS1) gene induce abnormal lysosomal alkalization, crippling the activity of resident hydrolases 3. Consequently, even if upstream phagophore formation is successful, the terminal fusion and degradation steps fail. This lysosomal bottleneck results in the pathological accumulation of undegraded mitophagosomes and massive intracellular inclusion bodies, physically choking neuronal architecture 3.
Amyotrophic Lateral Sclerosis and the TBK1-OPTN Axis
Advanced mechanistic research has solidified the centrality of mitophagy failure in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). These devastating motor neuron diseases are frequently linked to mutations in TANK-binding kinase 1 (TBK1) and the autophagy adaptor Optineurin (OPTN) 202122.
During a healthy mitophagy response, TBK1 physically associates with the OPTN adaptor at the site of the damaged mitochondrion. This physical interaction allows TBK1 to undergo auto-phosphorylation and subsequently phosphorylate OPTN at Serine 177 2021. The phosphorylation of Ser177 dramatically amplifies OPTN's binding affinity for LC3-II, firmly locking the ubiquitinated mitochondrion to the engulfing autophagosome 2021. Beyond OPTN, active TBK1 phosphorylates RAB7A to recruit membrane-donating ATG9 vesicles and phosphorylates p62 to further stimulate cargo capture 202523.
Over 90 distinct ALS/FTD-linked mutations have been identified within the TBK1 gene 2023. While some nonsense mutations result in simple haploinsufficiency, complex missense mutations offer profound mechanistic insights. For example, the E696K mutation located in TBK1's C-terminal domain leaves its catalytic kinase activity entirely intact but specifically abolishes its physical interaction with OPTN 21. Motor neurons derived from induced pluripotent stem cells (iPSCs) of patients with these mutations demonstrate a profound inability to form mitophagic rings around damaged cargo 2122. Consequently, these neurons exhibit a rapid accumulation of morphologically abnormal mitochondria alongside toxic p62 and TDP-43 protein aggregates 2122. This data unequivocally demonstrates that the spatial architecture and multimeric assembly of the OPTN-TBK1 complex is an absolute prerequisite for motor neuron survival.
The Aging Debate: Upstream Driver or Downstream Consequence
While it is undisputed that mitochondrial decline is a fundamental hallmark of biological aging, the chronological relationship between chronological age and mitophagy flux remains a subject of intense scientific debate. Do cells age because mitophagy inherently fails, or does mitophagy fail due to broader, downstream systemic collapse?
The Classical Decline Hypothesis
The classical gerontological hypothesis posits that basal mitophagy activity steadily decreases with advancing age 24. Proponents of this model argue that the transcriptional expression of essential autophagy genes diminishes over time, crippling the cell's ability to identify and isolate fused, senescent mitochondria. Consequently, tissues suffer from progressive bioenergetic failure and oxidative stress 2328.
Evidence supporting this classical model is heavily derived from in vitro cellular senescence models and peripheral blood analyses. Studies comparing primary human cells versus immortalized cell lines indicate that immortalization downregulates mitophagy, while replicative senescence impairs it entirely 24. Ex vivo analysis of CD4+ T-lymphocytes extracted from older adults reveals clear morphological evidence of stalled mitophagy, with electron microscopy showing massive accruals of dysfunctional mitochondria trapped within autophagic compartments 25. This indicates a systemic failure of mitochondrial turnover that directly contributes to the blunted immune defense and immunosenescence characteristic of aging cohorts 25.
The Compensatory Increase and Lysosomal Bottleneck Paradigm
However, recent technological advancements enabling high-resolution, single-cell in vivo imaging have challenged the universality of the classical decline model. Utilizing advanced fluorescent reporter mice (such as the mito-QC model), researchers have mapped the dynamic landscape of mitochondrial turnover across intact mammalian organ systems 30.
Remarkably, these studies reveal that decreased mitophagy is not a general, pervasive hallmark of healthy aging 30. In several critical neural subsets - including microglia and cerebellar granule cells - mitophagy levels remain stable or steadily increase throughout the lifespan 830. In the hippocampus, longitudinal trajectories show an age-dependent increase in mitophagic flux up until middle age, followed by a decline in extreme old age that nevertheless remains well above the baseline observed in youth 30.
These in vivo findings suggest a paradigm shift: aging tissues actively upregulate mitophagy as a compensatory, homeostatic response to the escalating burden of ROS and mitochondrial damage 825. When cellular health finally collapses, it is often not due to a failure in mitophagy initiation, but rather an insurmountable downstream bottleneck in global proteostasis - specifically, lysosomal dysfunction. As lysosomes accumulate indigestible lipofuscin cross-links and lose their critical acidity over decades, autophagosome-lysosome fusion is physically blocked 325. Thus, the cell continues to target mitochondria for destruction, but lacks the functional incinerators required to complete the process.
Inflammaging and the cGAS-STING Axis
The consequences of stalled mitochondrial clearance extend far beyond localized energy deficits, driving systemic physiological decline through chronic, low-grade inflammation, an aging phenotype termed "inflammaging."
When the mitophagic machinery fails to isolate fractured mitochondria, fragments of mitochondrial DNA (mtDNA) inevitably leak into the cellular cytoplasm 826. Because mtDNA is circular and lacks histones, the cytoplasm's innate immune surveillance systems misidentify it as invading bacterial or viral DNA 48. This cytosolic mtDNA binds to and activates the cyclic GMP-AMP synthase (cGAS), which in turn triggers the stimulator of interferon genes (STING) pathway located on the endoplasmic reticulum 427.
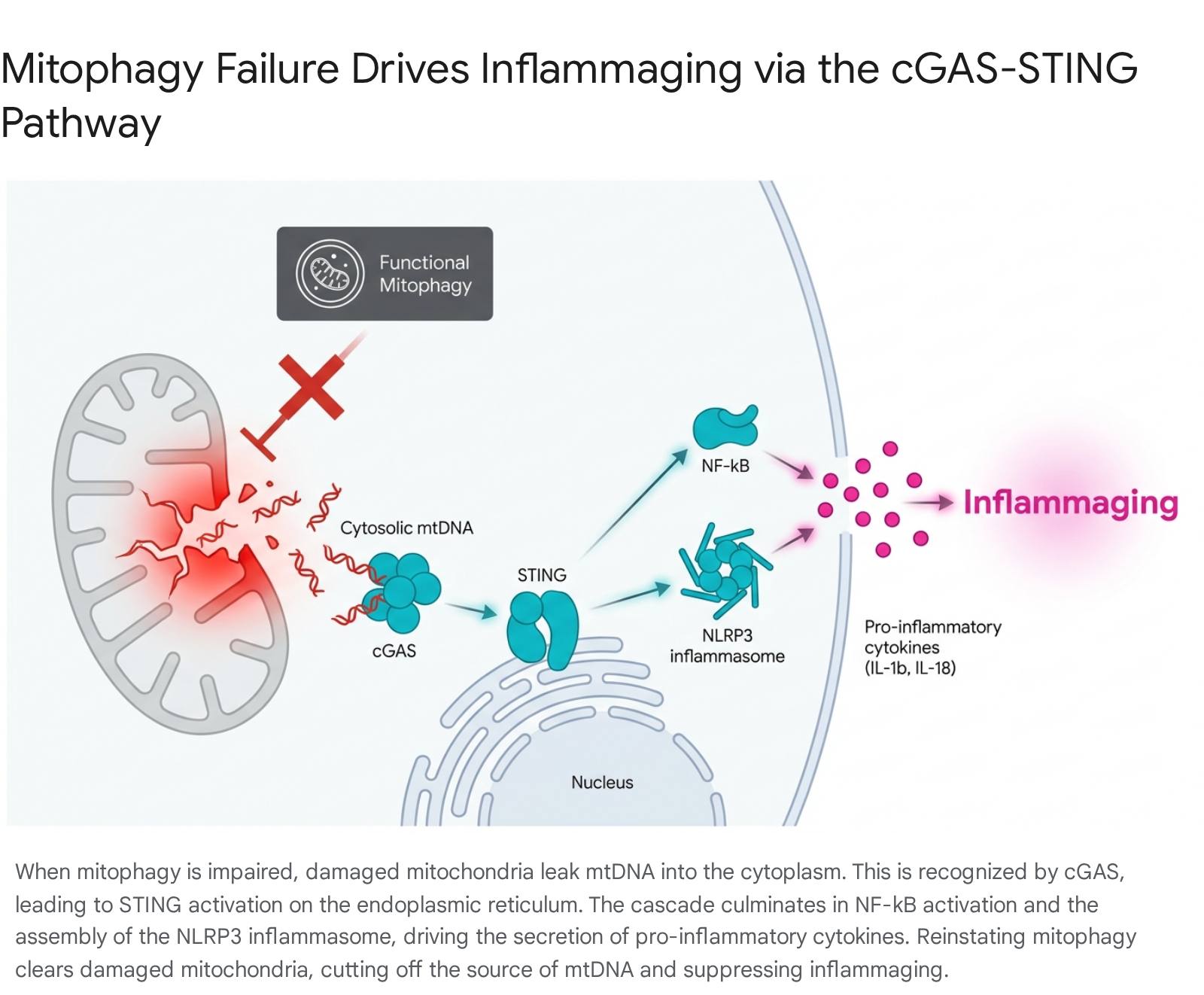
The activation of the cGAS-STING axis sets off a massive inflammatory cascade. It directly stimulates NF-κB and facilitates the assembly of the NLRP3 inflammasome, culminating in the secretion of potent pro-inflammatory cytokines, specifically IL-1β and IL-18 426. Prolonged secretion of these cytokines drives systemic tissue degradation, insulin resistance, and neuroinflammation. Transcriptomic analysis of aged tissues suggests that the observed compensatory increase in mitophagy is a deliberate cellular strategy deployed specifically to minimize mtDNA leakage and suppress cGAS-STING activation 8. Experimental restoration of mitophagy flux - either via PINK1 overexpression or pharmacological induction - rapidly clears immunogenic mtDNA, severs the cGAS-STING signaling loop, and reverses the inflammaging phenotype 427.
Pharmacological Modulators of Mitophagy
Recognizing that the enhancement of mitochondrial turnover can intercept both neurodegenerative decline and systemic aging, the targeted pharmacological induction of mitophagy has become a highly active area of drug development. Multiple distinct molecular classes are currently progressing through preclinical validation and human clinical trials.
Urolithin A
Urolithin A (UA) is a highly potent, naturally occurring metabolite derived from the degradation of dietary ellagitannins (found abundantly in pomegranates, walnuts, and berries) by the gut microbiome 283429. UA functions as a targeted mitophagy inducer by activating the AMPK pathway and upregulating PGC-1α, driving both the clearance of dysfunctional organelles and the compensatory biogenesis of new, healthy mitochondria 3037. Preclinical models of Alzheimer's disease show that UA supplementation significantly reduces Aβ plaque deposition, clears misfolded tau species, alleviates neuroinflammation, and restores the structural integrity of the blood-brain barrier 3437.
While dietary intake of ellagitannins is beneficial, its clinical utility is fundamentally limited by high inter-individual variability in gut microbiota. Only a subset of humans possesses the specific bacterial consortia capable of synthesizing UA. Cross-population analyses reveal that the "UM-A" and "UM-B" metabotypes (individuals capable of producing UA) account for roughly 40% to 55% of populations across America, Spain, and China 29. Conversely, 10% to 17% of individuals exhibit the "UM-0" metabotype, meaning they lack the microbial capacity to produce UA regardless of their diet 29. This high demographic variability strictly necessitates direct, standardized oral supplementation of UA to ensure consistent therapeutic efficacy.
Recent randomized, double-blind, placebo-controlled human clinical trials have demonstrated robust safety and profound systemic benefits following direct UA supplementation. The landmark 2025 MitoImmune trial published in Nature Aging investigated the administration of 1,000 mg of UA daily over 28 days in healthy adults aged 45 - 70 313932. Results indicated a systemic rejuvenation of the immune profile: UA significantly increased the proportion of naive CD8+ T cells, reduced markers of cellular exhaustion, and shifted immune cell metabolism away from glucose reliance toward more efficient fatty acid and amino acid oxidation 313932. Furthermore, UA supplementation successfully suppressed markers of inflammaging, including IL-6 and TNF 31. Additional trials, such as the ongoing CLARITY study (slated for completion in 2026 with 650 participants), are currently assessing the capacity of UA to protect against cognitive decline in large cohorts 3141. Other phase 2 studies are simultaneously evaluating its efficacy in preserving muscle mass during weight loss in obese adults (BMI >30) 33.
Spermidine
Spermidine is a naturally occurring polyamine abundant in wheat germ, aged cheeses, and soybeans. While UA specifically targets mitochondrial quality control, spermidine acts as a broad-spectrum inducer of general macroautophagy 2843. It exerts its geroprotective effects by inhibiting mTOR, activating the AMPK and SIRT1 pathways, and driving the hypusination of the translation initiation factor eIF5A, a highly conserved metabolic switch required for autophagic execution 162844.
Spermidine's ability to cross the blood-brain barrier makes it a particularly compelling candidate for neuroprotection 4534. In aged murine models, dietary supplementation enhances spatial navigation and object recognition memory, while significantly protecting neurons from apoptotic cell death 2845. Retrospective epidemiological data corroborate these findings in humans, where higher dietary spermidine intake is robustly correlated with reduced all-cause mortality, diminished biological aging, and lower incidences of Alzheimer's disease and severe dementia 35.
To move beyond epidemiology, large-scale clinical interventions have tested direct spermidine supplementation across diverse demographic cohorts, including substantial trials within Asian populations (e.g., Japan and China), contributing to a more globally representative dataset 363738. Interventional trials administering daily spermidine to older adults suffering from subjective cognitive decline (SCD) or mild-to-moderate dementia have shown excellent safety profiles and measurable, dose-dependent improvements in memory performance and cognitive plasticity over a 12-month period 453435.
Alpha-Ketoglutarate and NAD+ Precursors
- Alpha-Ketoglutarate (AKG): AKG is a fundamental intermediate metabolite of the tricarboxylic acid (TCA) cycle. Beyond energy metabolism, AKG extends lifespan and severely delays morbidity in aging models by acting as a caloric restriction mimetic. It binds directly to and inhibits mitochondrial ATP synthase, which slightly reduces ATP levels and signals a starvation response that suppresses mTOR and upregulates autophagy and mitophagy 3940. Extensive murine studies demonstrate that dietary AKG preserves skeletal muscle mass, improves gait, reduces tumor incidence, and results in a dramatic compression of late-life morbidity 41. Clinical trials assessing its efficacy on epigenetic clocks and physiological decline in middle-aged humans (45 - 65 years) are actively underway 41.
- NAD+ Precursors: The bioavailability of nicotinamide adenine dinucleotide (NAD+) declines significantly with age, impairing sirtuin function and mitochondrial bioenergetics. Supplementation with biosynthetic precursors, primarily nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN), effectively replete intracellular NAD+ pools 2142. Replenishing NAD+ rescues neuronal survival by countering the hyperactivation of NAD-consuming enzymes during autophagic stress, preventing subsequent mitochondrial depolarization 21. Multiple clinical trials have confirmed that oral doses of NMN (e.g., 250 mg) and NR (e.g., 1,000 mg) are safe, elevate whole-blood NAD+ levels, and improve insulin sensitivity and lower limb function in elderly cohorts, though human efficacy often appears less pronounced than in preclinical rodent models 42554357.
| Compound / Molecule | Primary Biological Target | Primary Mechanism of Action | Clinical / Phenotypic Highlights |
|---|---|---|---|
| Urolithin A | Targeted Mitophagy | Activates AMPK, upregulates PGC-1α; facilitates direct clearance of damaged mitochondria. | Improves muscle endurance; rejuvenates CD8+ T cells; strongly reduces systemic inflammaging. |
| Spermidine | General Macroautophagy | mTOR inhibition, AMPK/SIRT1 activation, essential hypusination of eIF5A. | Prevents age-related cognitive decline; enhances memory performance in clinical dementia trials. |
| Alpha-Ketoglutarate | Energy Metabolism / Autophagy | Direct inhibition of ATP synthase and mTOR; acts as a potent caloric restriction mimetic. | Extends overall lifespan; achieves dramatic compression of late-life morbidity in mammalian models. |
| NAD+ Precursors | Mitochondrial Bioenergetics | Repletes systemic NAD+ pools to support sirtuin activity and metabolic homeostasis. | Demonstrates high clinical safety; improves insulin sensitivity and provides broad cardiovascular protection. |
| MIC (Coumarin) | Lysosome & Mitophagy | TFEB inducer via DAF-12/FXR inhibition. | Drives simultaneous activation of lysosomal biogenesis and mitochondrial clearance. |
Advancements in Mitophagy Measurement and Biomarkers
Historically, the evaluation of autophagic and mitophagic flux relied upon cumbersome, low-throughput techniques. Electron microscopy provided excellent spatial resolution of engulfed mitochondria but lacked quantitative scalability, while static western blotting of mitochondrial membrane proteins was often confounded by concurrent proteasomal degradation 4445. The inability to capture the dynamic, temporal flux of mitochondrial turnover posed a significant bottleneck for translational aging research.
In Vitro and Ex Vivo Fluorescent Reporters
Significant methodological leaps have been achieved with the engineering of advanced, pH-sensitive fluorescent fusion proteins. The mt-Keima reporter consists of a ratiometric fluorescent protein targeted specifically to the mitochondrial matrix via the COX8 sequence 4445. Because its excitation spectrum shifts dramatically depending on environmental pH, healthy mitochondria at a neutral pH (7.0 - 8.0) emit green fluorescence, whereas mitochondria that have been successfully engulfed by highly acidic autolysosomes (pH ~4.5) emit red fluorescence. This colorimetric shift allows researchers to rapidly quantify active, completed mitophagy flux in living cells and tissues 4446.
Similarly, the mito-QC assay utilizes a tandem mCherry-GFP tag fused to the mitochondrial targeting sequence of the OMM protein FIS1 545. Because the GFP signal is rapidly quenched by acidic environments while the mCherry signal remains stable, healthy mitochondrial networks appear yellow (mCherry+GFP+), while mitochondria undergoing degradation appear as exclusively red puncta (mCherry+GFP-) 4547. Recent advancements detailed in late 2024 literature describe integrating the mito-QC reporter with spectral flow cytometry. This combination creates highly quantitative, medium-throughput platforms capable of multiplexing mitophagy assessment alongside cellular viability and ROS production probes, accelerating the discovery and validation of novel mitophagy modulators 474849.
Clinical Translation: PET Tracers in Human Neurology
While fluorescent reporters are transformative for preclinical animal models, tracking mitochondrial pathology and neurodegeneration in living human patients relies entirely on advanced neuroimaging. The field of radiopharmaceuticals has experienced explosive, unprecedented growth, with the US FDA approving 21 novel positron emission tomography (PET) tracers between 2020 and late 2023 50.
Crucially, the quest to visualize the downstream neuropathological consequences of mitophagy failure - specifically, the toxic aggregation of alpha-synuclein in Parkinson's disease and Multiple System Atrophy (MSA) - is now approaching clinical viability. Developing a highly specific α-synuclein PET tracer has historically been fraught with difficulties due to off-target binding to structurally similar amyloid-beta and tau aggregates 19. However, breakthrough candidate compounds presented globally in 2024 and 2025, such as [11C]M503-1619, have largely overcome these hurdles.
Identified through ultrahigh-throughput in silico screening of over 42 million compounds, [11C]M503-1619 demonstrates a remarkably high binding affinity (Ki = 6.5 nM) to specific residues (site 9) of α-synuclein fibrils, while displaying negligible affinity for beta-amyloid 5152. In non-human primate models, this radioligand exhibits excellent brain uptake and rapid washout, essential characteristics for high-contrast PET imaging 52. Alongside other emerging candidates like [18F]ACI-12589, these targeted PET tracers are currently undergoing rigorous human testing. Their successful deployment in clinical settings represents a monumental step toward diagnosing mitophagy-related neurodegeneration at its earliest stages, allowing for precise monitoring of therapeutic interventions aimed at restoring autophagic flux 1951.
Conclusion
Mitophagy constitutes an indispensable pillar of cellular homeostasis, functioning at the precise intersection of energy metabolism, proteostasis, and innate immunity. The molecular orchestration of this pathway - ranging from AMPK-driven initiation and PINK1/Parkin-mediated ubiquitin tagging to the final execution of lysosomal degradation - protects specialized, post-mitotic cells against the toxic sequelae of cumulative oxidative stress.
As the global population ages, the clinical consequences of unmitigated mitochondrial dysfunction are starkly visible in the rising incidence of devastating neurodegenerative diseases, including Alzheimer's, Parkinson's, and ALS. Evolving transcriptomic and in vivo imaging evidence continues to reshape our understanding of biological aging, strongly suggesting that systemic "inflammaging" is deeply intertwined with the cell's failure to contain mtDNA leakage via mitophagic clearance. Promisingly, the rapid development of highly specific measurement tools and the successful progression of potent, targeted mitophagy inducers - such as Urolithin A and Spermidine - into advanced clinical trials offer a realistic horizon for disease-modifying therapies. By pharmaceutically restoring the autolysosomal degradation of damaged mitochondria, modern geromedicine holds the profound potential to compress late-life morbidity, suppress chronic inflammation, and preserve neurological integrity well into old age.