Mitochondrial heteroplasmy and its role in aging
Biological Mechanisms of Mitochondrial Genetics
Mitochondria are semi-autonomous organelles responsible for generating the vast majority of cellular adenosine triphosphate (ATP) through the process of oxidative phosphorylation (OXPHOS). Arising from an ancient endosymbiotic event in which an ancestral eukaryotic cell engulfed an $\alpha$-proteobacterium, mitochondria maintain their own distinct circular genome, known as mitochondrial DNA (mtDNA) 12. In humans, this compact 16.5-kilobase genome is notably dense, lacking introns and coding for exactly 37 genes. These comprise 13 essential polypeptide subunits of the OXPHOS electron transport chain (ETC), 22 transfer RNAs (tRNAs), and two ribosomal RNAs (rRNAs) 134. The synthesis of a fully functional OXPHOS system requires complex nuclear-mitochondrial genome coordination, as over 75 to 1,000 nuclear-encoded proteins must be imported into the mitochondria to maintain structural integrity, regulate genome replication, and assemble the respiratory complexes 456.
Unlike the diploid nuclear genome, which exists in two copies per cell, mtDNA is present in hundreds to thousands of copies per cell, governed by the specific energy demands of the host tissue 78. Because mtDNA replicates independently of the cell cycle and resides within the mitochondrial matrix in close proximity to the primary cellular source of reactive oxygen species (ROS) - the electron transport chain - it is subject to an accelerated mutation rate. Estimates indicate that the mtDNA mutation rate is 10 to 100 times higher than that of nuclear DNA, compounded by the absence of protective histone packaging and the reliance on less comprehensive DNA repair mechanisms 91011.
Because cells contain multiple copies of the mitochondrial genome, a newly mutated mtDNA molecule can coexist alongside the wild-type (normal) mtDNA within the same cell or even the same organelle. This state of genomic admixture is termed heteroplasmy 812. The ratio of mutated to wild-type mtDNA, referred to as the variant allele frequency (VAF) or heteroplasmic load, exhibits profound variability among different tissues, among different cells within the same tissue, and even among individual mitochondria within a single cell 813. The dynamic fluctuations in heteroplasmy over time drive age-related functional decline, as somatic mutations accumulate, clonally expand, and eventually impair cellular bioenergetics.
Accumulation of Somatic Mutations in Aging
The progressive accumulation of mtDNA mutations is universally recognized as a primary hallmark of cellular aging. However, the precise mechanisms governing how rare, stochastic mutations arise and expand to high intracellular frequencies have been subject to continuous evolution in the scientific literature.
The Vicious Cycle Hypothesis
For decades, the prevailing paradigm explaining age-associated mtDNA mutation accumulation was the "vicious cycle" hypothesis. This model suggested that initial oxidative damage to mtDNA impairs the efficiency of the OXPHOS machinery, leading to increased leakage of electrons from the transport chain 1415. This leakage inevitably produces elevated localized levels of reactive oxygen species, which subsequently inflict further damage upon the remaining mtDNA molecules. The theory proposed a positive feedback loop of accelerating genomic damage and escalating functional decline 1516.
However, advanced molecular tracking has largely challenged the vicious cycle as the exclusive driver of heteroplasmic expansion. Studies utilizing ultra-accurate Duplex Sequencing reveal that mutations associated with oxidative damage rarely form dominant clones in aged tissues 17. Furthermore, administering targeted antioxidants to aging animal models has resulted in the clearance of certain oxidative lesions, suggesting a dynamic lifelong clearance process rather than a strictly cumulative, runaway cycle of oxidative destruction 17.
Random Genetic Drift and Clonal Expansion
Contemporary models of mtDNA mutation accumulation heavily favor the concept of random genetic drift, particularly within post-mitotic tissues such as skeletal muscle fibers and central nervous system neurons. Foundational computer simulations and single-cell molecular studies have demonstrated that the clonal expansion of a single mtDNA mutation can occur entirely through relaxed replication and continuous organelle turnover 21418.
In this model, mtDNA molecules are continually replicated and degraded independently of the broader cellular division cycle. Over the course of decades, random sampling during these continuous turnover events can inadvertently lead to the expansion of a single mutant allele. This process, governed by stochastic drift, allows an initially singular mutation to multiply and dominate the intracellular environment without conferring any inherent replicative advantage to the organelle 214. Mathematical modeling indicates that this random drift is sufficient to explain the decades-long latency observed before specific cells, such as those in the human colorectal epithelium, exhibit complete cytochrome c oxidase (COX) deficiency 218. In vivo studies utilizing heterozygous PolgA 'mutator' mice corroborate this framework, demonstrating that while the absolute level of de novo mtDNA mutations remains constant, existing mutations progressively expand to higher heteroplasmy levels, leading to age-associated respiratory chain dysfunction 19.
Positive Selection of Deleterious Variants
While random genetic drift explains clonal expansion in non-dividing tissues, recent large-scale longitudinal studies have introduced evidence for the positive selection of deleterious mtDNA variants at the cellular level.
Data derived from 2024 longitudinal studies analyzing somatic heteroplasmy in prospective human cohorts over an 8.6-year period revealed that heteroplasmies predicted to be deleterious were statistically more likely to increase in variant allele frequency over time 202122. This directional accumulation challenges the assumption that somatic mtDNA mutations in aging arise and expand solely due to neutral drift. The data indicate that certain mutations - despite exerting a negative impact on overall organismal mortality - confer a competitive replicative advantage to the individual mitochondrial genomes that harbor them, resulting in a localized "destructive" selection that inherently favors non-functional genomes 1623.
Further supporting this multi-level selection dynamic, large-scale genomic analyses utilizing data from over 750,000 individuals identified a two-step mechanism linking mtDNA mutations to systemic aging phenomena, such as clonal hematopoiesis. Initially, individual cells randomly accumulate low levels of cryptic mtDNA mutations. When a specific cell clone gains a proliferative advantage and expands - often driven by distinct somatic mutations in nuclear genes such as TERT, TCL1A, or SMC4 - the cryptic mtDNA variants are carried along as passenger mutations, becoming highly detectable in whole blood analyses 24. This paradigm demonstrates that mtDNA heteroplasmy in highly proliferative tissues is inextricably linked to the selective pressures and proliferative dynamics operating on the nuclear genome.
The Biochemical Threshold Effect
The presence of mutated mtDNA within a cell does not immediately result in overt metabolic dysfunction. Because multiple wild-type mitochondrial genomes can complement and compensate for defective ones, a critical intracellular ratio must be reached before cellular respiration is meaningfully impaired. This functional tipping point is defined in the literature as the biochemical threshold effect 1325.
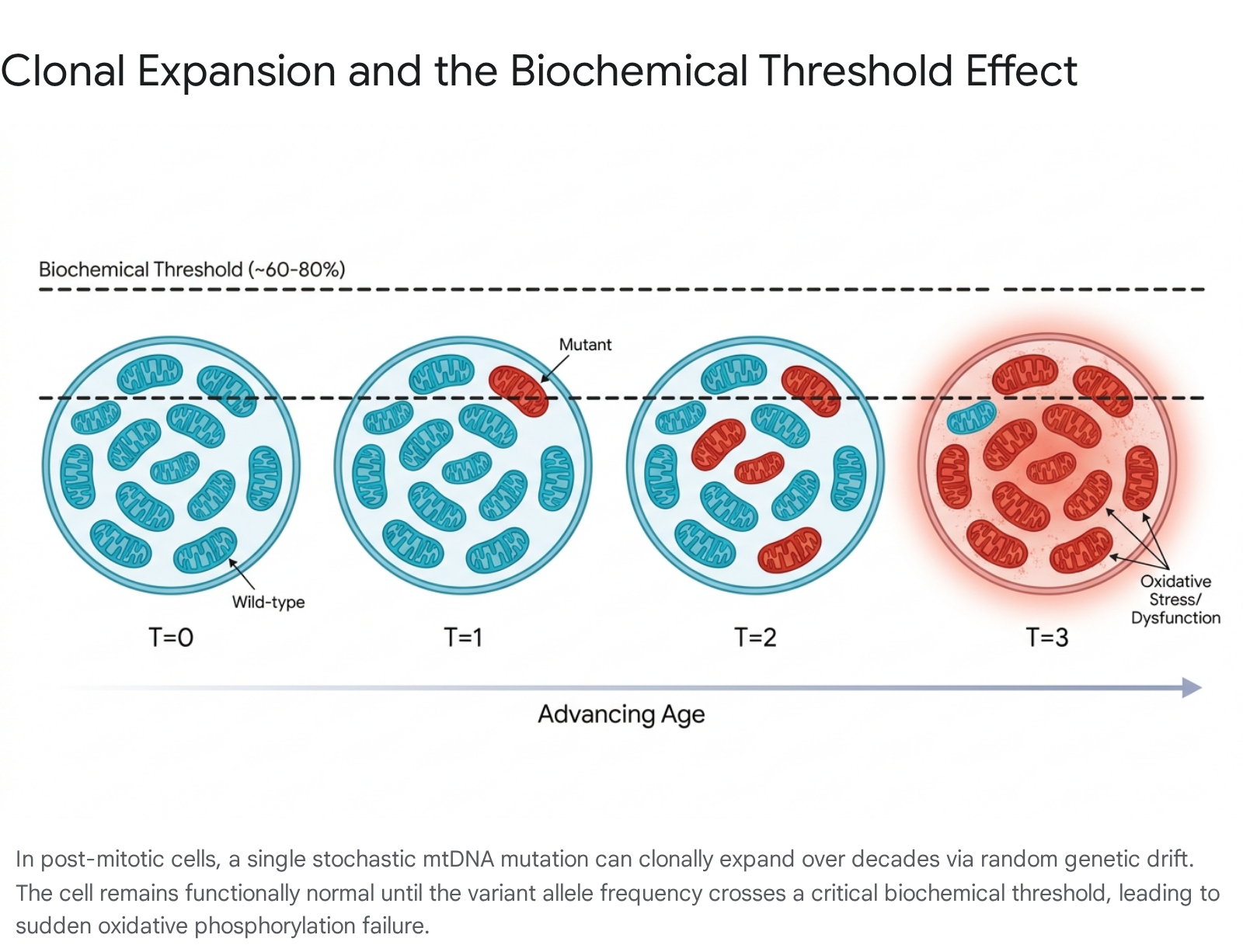
Variability of Mutation Thresholds
The specific variant allele frequency required to induce a measurable oxidative phosphorylation defect generally ranges from 60% to 90% mutant load. However, this figure is highly variable and depends intrinsically on the specific nature of the mutation, the affected mitochondrial gene, and the overarching energetic reliance of the host tissue 252627.
An important distinction exists between the biochemical threshold at the isolated cellular level - beyond which measurable mitochondrial dysfunction occurs in individual cells - and the clinical threshold in bulk organs, above which the collective impairment of multiple cells results in overt systemic symptoms 28. Certain single nucleotide variants (SNVs), such as the m.8993T>G mutation affecting the MT-ATP6 gene, demonstrate a direct correlation with diminished complex V activity in skeletal muscle once the variant allele frequency surpasses roughly 60% 1327. Conversely, in highly proliferative cells like fibroblasts, the correlation between m.8993T>G VAF and respiratory impairment is statistically non-significant, reflecting the differing metabolic dependencies of the cell types 1327.
Tissue-Specific Bioenergetics and Vulnerabilities
Systematic bioenergetic analyses demonstrate significant variance in the stoichiometric ratios of respiratory complexes across different human tissues, dictating the distinct vulnerabilities of each organ. The structural ratios of complexes II, III, and IV differ markedly between the liver, kidney, skeletal muscle, and brain, determining how much "spare respiratory capacity" each tissue maintains 4.
Highly energetic, post-mitotic tissues such as the brain (neurons), the heart (cardiomyocytes), and skeletal muscle rely almost entirely on continuous OXPHOS for survival 102930. These tissues are exceptionally sensitive to OXPHOS impairments and exhibit lower biochemical thresholds for disease manifestation. Conversely, tissues with higher metabolic plasticity, which can rapidly upregulate cytosolic glycolysis to compensate for mitochondrial deficits, can tolerate significantly higher heteroplasmic loads before exhibiting pathology 13313233.
Table 1 summarizes the relative impact and threshold characteristics of heteroplasmic accumulation across critical tissue types.
| Tissue Type | Primary Energy Reliance | Common Age-Related mtDNA Alterations | Typical Threshold for OXPHOS Defect | Phenotypic Consequences of Exceeding Threshold |
|---|---|---|---|---|
| Brain (Neurons) | Extreme (Strict continuous OXPHOS) | Large-scale structural deletions, point mutations | ~60% - 70% | Axonal degeneration, stroke-like episodes, cognitive decline, neuronal apoptosis 343536. |
| Cardiac Muscle | High (Predominantly fatty acid oxidation) | Point mutations, progressive accumulation of "common deletion" | ~60% - 80% | Hypertrophic cardiomyopathy, arrhythmias, compromised intracellular calcium handling 373839. |
| Skeletal Muscle | High (Mixed OXPHOS and glycolysis) | Large-scale deletions, m.8993T>G | ~60% - 80% | Sarcopenia, presence of ragged-red fibers, decreased maximal oxygen consumption (VO2max) 62740. |
| Liver / Epithelium | Moderate (High metabolic plasticity) | Assorted point mutations (clonally expanded in stem cell niches) | >80% - 90% | Hepatic dysfunction; generally exhibits higher tolerance to elevated mutation loads 418. |
Clinical Dynamics of the m.3243A>G Mutation
The clinical complexity of the biochemical threshold is prominently illustrated by the m.3243A>G mutation, an A-to-G transition in the MT-TL1 gene encoding the mitochondrial tRNA for leucine 313441. This mutation is the most frequent pathogenic mtDNA variant in humans and is the primary driver of Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes (MELAS), as well as Maternally Inherited Diabetes and Deafness (MIDD) 263134.
Research utilizing induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) from patients harboring the m.3243A>G mutation reveals profound downstream metabolic divergence. When heteroplasmic loads exceed 70% to 80%, severe respiratory chain defects reliably manifest. However, the subsequent cellular response varies: some patient-derived cells successfully undergo a compensatory upregulation of glycolysis, thereby maintaining stable intracellular ATP levels. Other isogenic lines fail to execute this metabolic shift, resulting in severe ATP depletion, altered calcium signaling, and the eventual development of hypertrophic cardiomyopathy 3137.
In the central nervous system, high heteroplasmy of the m.3243A>G mutation generates highly specific vulnerabilities within long-range projection neurons. Deep-layer cortical neurons subjected to mitochondrial stress undergo rapid axonal degeneration and apoptosis, closely mirroring the post-mortem neuropathology of MELAS patients 34. Furthermore, longitudinal studies assessing m.3243A>G heteroplasmy in blood leukocytes demonstrate a phenomenon of purifying selection within mitotic tissues, wherein the variant allele frequency undergoes a biphasic decline of approximately 1.4% to 2.3% per year as highly mutated hematopoietic lineages are outcompeted by healthier clones 4142.
Transgenerational Transmission and Purifying Selection
In direct contrast to the uncontrolled somatic accumulation of mutations during organismal aging, the female germline deploys rigorous genetic quality control mechanisms to prevent the transgenerational inheritance of deleterious mtDNA.
The Mitochondrial Bottleneck
During embryonic development, mammalian reproductive biology utilizes a "mitochondrial bottleneck," a process that drastically reduces the absolute number of mtDNA copies present in primordial germ cells 2843. This physical reduction forces rapid genomic segregation, ensuring that heteroplasmy levels diverge significantly between different oocytes.
Recent murine models indicate that the stringency of this bottleneck correlates directly with genetic health. When fewer mitochondrial DNA copies are passed from mother to offspring, there is greater genetic variation between individual ova, allowing purifying selection to operate with enhanced efficiency 43. This selective mechanism actively identifies and eliminates defective mtDNA lineages, preventing a multi-generational mutational meltdown 4344. When researchers experimentally impaired cellular autophagy - disrupting the cell's ability to recycle damaged organelles - the protective filtering of the bottleneck weakened, resulting in a precipitous decline in transgenerational mtDNA quality 43.
The Impact of Maternal Age
The relationship between maternal age and mtDNA mutation transmission has historically been modeled on the assumption that older oocytes accumulate and transmit higher heteroplasmic loads. However, precision genome editing studies utilizing DddA-derived cytosine base editors (DdCBEs) to create highly specific pathogenic mutations in mouse complex I genes (ND1 and ND5) have challenged this paradigm.
Recent findings published in 2024 reveal that both specific mutations remain under intense purifying selection during maternal transmission, a process occurring predominantly during postnatal oocyte maturation 45. Paradoxically, the data demonstrate that advanced maternal age actively enhances this purifying selection. The older maternal environment appears to offer a protective metabolic effect against the transmission of deleterious mtDNA mutations, suggesting that aged oocytes may enforce more rigorous metabolic checkpoints during final maturation stages 45.
Cellular Reprogramming and Aging Phenotypes
The intricate relationship between mitochondrial heteroplasmy and the biological aging process is distinctly observable during cellular reprogramming. When differentiated somatic cells are subjected to specific transcription factors to generate induced pluripotent stem cells (iPSCs) - an intervention meant to reverse cellular epigenetic age and reinstate pluripotency - the mitochondrial genome exhibits highly variable segregation behaviors.
Heteroplasmy in Induced Pluripotent Stem Cells
A pivotal 2024 study reprogrammed human fibroblast lines harboring known heteroplasmies for deleterious mtDNA point and deletion mutations. The quantification of mutation heteroplasmy across 76 resulting iPSC clones revealed a starkly bimodal distribution. Following the reprogramming bottleneck, specific clones successfully cleared the mutant genomes, dropping to near-zero heteroplasmy, while other clones experienced massive clonal expansion, stabilizing at exceptionally high mutation levels 4647.
The downstream consequences of this segregation provide direct evidence linking mtDNA heteroplasmy to fundamental aging phenotypes. The iPSC clones burdened with elevated deletion mutation heteroplasmy exhibited profound functional impairments. Transcriptomic and metabolic profiling of these highly mutated clones demonstrated a significant metabolic shift toward neutral lipid synthesis and altered growth dynamics 4647. More critically, the cells harboring elevated heteroplasmy exhibited a markedly increased epigenetic age compared to their isogenic, mutation-free counterparts 4647. This establishes that high mtDNA mutation heteroplasmy is not merely a correlative bystander of advancing chronological age but a direct, active inducer of cellular senescence and aging phenotypes.
The Mitochondrial Unfolded Protein Response
The acquisition of pluripotency and the determination of cell fate are heavily influenced by retrograde signaling from the mitochondria to the nucleus. The mitochondrial unfolded protein response (UPRmt) serves as a primary regulatory pathway in this dialogue. Various stressors, including imbalances in mitochondrial protein homeostasis, elevated reactive oxygen species, and overt OXPHOS dysfunction, trigger the UPRmt 48.
Recent investigations detail that the UPRmt is transiently activated during the early stages of acquiring pluripotency but must gradually diminish to allow normal development. Persistent activation of the UPRmt - acting through transcription factors such as c-Jun - reduces intracellular acetyl-CoA levels, thereby decreasing histone acetylation. This epigenetic alteration inhibits the mesenchymal-to-epithelial transition (MET), effectively hindering the acquisition of pluripotency 48. This mechanism underscores how the energetic status and genomic integrity of the mitochondria continuously dictate the nuclear epigenetic landscape during differentiation and aging.
Methodologies for Heteroplasmy Detection
Historically, the ability to characterize the precise architecture of mitochondrial heteroplasmy across human tissues was constrained by the technical limitations of conventional sequencing platforms. Traditional Sanger sequencing and early iterations of next-generation sequencing (NGS) of bulk tissue homogenates frequently masked the true complexity of aging biology, leading to debates regarding the true prevalence of somatic mutations.
Limitations of Bulk Sequencing
Bulk RNA and DNA sequencing methodologies rely on homogenizing thousands to millions of cells to extract pooled genetic material. Consequently, this process averages out the mutational signals across the entire sample 495050. If a severe, pathogenic mtDNA deletion is present at 100% homoplasmy in a single degenerating cell, but completely absent in the surrounding thousands of healthy support cells, bulk sequencing algorithms will report an overall heteroplasmic load of a fraction of a percent. This signal is virtually indistinguishable from the background sequencing error rate of standard Illumina short-read platforms, which typically hover between 0.1% and 1.0% 51525354.
Furthermore, bulk analyses are exceptionally vulnerable to contamination from nuclear mitochondrial DNA-like sequences (NUMTs). Over millions of years of evolutionary history, fragmented segments of mitochondrial DNA have naturally migrated and inserted themselves into the nuclear genome. When short-read bulk sequencing is utilized, these static nuclear pseudogenes are frequently amplified and misidentified as genuine heteroplasmic mtDNA mutations. This technical artifact can artificially inflate mutation counts, heavily confounding the interpretation of aging data 71254. Systematic evaluations indicate that adjusting alignment protocols - such as mapping exclusively against the revised Cambridge Reference Sequence (rCRS) versus mapping against the whole human genome (GRCh38) to trap NUMTs - can drastically alter the detection of false-positive heteroplasmies 754.
Single-Cell Sequencing Approaches
To resolve the exact cellular architecture of heteroplasmy, geneticists have increasingly turned to ultra-sensitive single-cell profiling. Single-cell RNA sequencing (scRNA-seq) and droplet-based mitochondrial single-cell Assay for Transposase-Accessible Chromatin with sequencing (mtscATAC-seq) provide unparalleled cellular resolution.
These platforms allow researchers to trace the lineage and clonal expansion of individual cells in vivo by utilizing native somatic mtDNA mutations as highly reliable natural barcodes 81151. Because the mitochondrial genome exists in high copy numbers, retrospective inference of cellular relationships via mtDNA mutations is significantly more cost-effective and robust than attempting mutation detection in the vast nuclear genome 11. Moreover, techniques like mtscATAC-seq enable the simultaneous profiling of the accessible chromatin landscape alongside high-confidence mtDNA mutation calling. This dual-omics approach allows researchers to definitively link a specific heteroplasmic state to precise transcriptomic and epigenetic alterations within an individual cell 811.
Duplex Sequencing and Individual Molecule Techniques
To address the intrinsic base-calling error rates of standard NGS protocols, researchers deploy Duplex Sequencing. This technique attaches randomized, double-stranded molecular barcodes to both strands of an individual DNA molecule prior to PCR amplification. For a mutation to be classified as genuine, it must be independently read on both the forward and reverse strands of the original source molecule, allowing computational algorithms to systematically eliminate stochastic PCR artifacts and sequencing machine errors 91753. Duplex Sequencing reduces the error floor to less than 0.01% VAF, revealing that aging mammalian tissues accumulate highly specific classes of point mutations at predictable, clock-like rates 95253.
Despite its accuracy for single nucleotide variants (SNVs), Duplex Sequencing requires heavy DNA fragmentation, destroying critical haplotype linkage information. To circumvent this, the field has developed Individual Mitochondrial Genome sequencing (iMiGseq). This long-read technique avoids both amalgamation and fragmentation by sequencing full-length individual mtDNA molecules. It enables the detection of ultra-rare variants, resolves complete structural haplotypes without NUMT interference, and provides precise quantification of sequentially acquired, linked heteroplasmic mutations on the same physical genome 5355.
Table 2 outlines the analytical characteristics and limitations of current mtDNA sequencing platforms.
| Sequencing Methodology | Analytical Resolution | Primary Technical Advantage | Key Limitations and Constraints | Approximate Error Rate / Minimum Detection Limit |
|---|---|---|---|---|
| Bulk NGS (Short-read) | Tissue-average | Highly cost-effective; robust for identifying broad population-level homoplasmic trends. | Completely masks cellular heterogeneity; highly susceptible to NUMT interference and amplification bias 7849. | ~1.0% to 2.0% VAF 5356. |
| Single-Cell (mtscATAC-seq) | Individual Cellular | Links heteroplasmy directly to specific epigenetic cell states; enables in vivo clonal lineage tracing 1151. | High dropout rates; elevated per-sample sequencing costs; variable coverage depth across genome 850. | ~1.0% to 3.0% VAF (highly dependent on read depth) 57. |
| Duplex Sequencing | Molecular | Ultra-high accuracy; computationally eliminates PCR and base-calling bias 953. | Destroys critical haplotype linkage information due to mandatory DNA fragmentation 5355. | < 0.01% VAF 5253. |
| iMiGseq (Long-read) | Molecular (Full Genome) | Resolves complete haplotypes; identifies co-occurring structural variants on a single physical molecule 5355. | Lower overall throughput than short-read methods; requires highly specialized bioinformatics pipelines. | < 0.005% VAF 53. |
Therapeutic Interventions Targeting Mitochondrial Dysfunction
As the causal molecular mechanisms linking mitochondrial heteroplasmy to organismal aging are further elucidated, clinical research has pivoted toward therapeutic interventions capable of either artificially clearing defective mitochondria or directly editing the mutant mitochondrial genomes in vivo.
Targeted Mitophagy Enhancers
Mitophagy is a highly selective form of macroautophagy responsible for identifying, sequestering, and degrading dysfunctional or depolarized mitochondria. This quality control mechanism is primarily governed by the PINK1/Parkin ubiquitin signaling pathway, alongside ubiquitin-independent receptor pathways involving proteins like BNIP3 and FUNDC1 585960. In youthful cellular environments, robust mitophagy effectively identifies and clears organelles harboring elevated levels of mutant mtDNA. During normal aging, the efficiency of this autophagic quality control system declines sharply, allowing heteroplasmic loads to escape containment, cross biochemical thresholds, and trigger a cascade of senescence-associated secretory phenotypes (SASP) 596061.
The pharmacological restoration of mitophagy represents a premier anti-aging strategy entering clinical development. Urolithin A (UA), a postbiotic metabolite synthesized by the gut microbiome from dietary polyphenols (ellagitannins), has been identified as a potent, targeted mitophagy activator. By stimulating the AMPK signaling pathway, UA prompts the selective degradation of defective mitochondria while preserving functional networks 626364. Recent preclinical and clinical studies conducted in 2024 and 2025 demonstrate that oral UA supplementation can significantly reduce markers of systemic inflammation (such as C-reactive protein), improve aerobic endurance, and increase measurable skeletal muscle strength by up to 12% in middle-aged adult cohorts 296263.
Similarly, next-generation therapeutics focus on restoring cellular NAD+ levels utilizing precursors like Nicotinamide Mononucleotide (NMN) or Nicotinamide Riboside (NR) to enhance sirtuin-mediated (SIRT1/SIRT3) mitophagy pathways 596064. Concurrently, pharmaceutical development of highly specific deubiquitylating enzyme inhibitors, such as Mission Therapeutics' USP30 inhibitors (MTX652 and MTX325), aims to remove the biological "brakes" on the mitophagy process, promoting the accelerated clearance of dysfunctional mitochondria in neurodegenerative diseases like Alzheimer's and Parkinson's 65.
Despite these promising mechanisms, researchers caution that metabolic interventions possess inherent limitations. Because interventions relying on metabolic plasticity - including physical exercise, dietary restriction, and specific pharmaceuticals - demand robust levels of baseline ATP to execute cellular remodeling, they may exhibit diminished efficacy in advanced older age when severe mitochondrial bioenergetic dysfunction is already deeply established 33.
Mitochondrial Antioxidants
While untargeted systemic antioxidants have historically failed to alter aging trajectories, mitochondrially-targeted antioxidants represent a refined approach. Compounds such as MitoQ and SkQ1 are engineered to accumulate specifically within the mitochondrial matrix, directly neutralizing the highly reactive oxygen species generated by heteroplasmic OXPHOS dysfunction at their origin 6466. Current clinical trials (e.g., NCT03514875) are investigating the capacity of MitoQ to reduce localized endothelial oxidative stress, thereby increasing nitric oxide bioavailability and improving cerebrovascular blood flow in patients suffering from mild cognitive impairment 66.
Precision Mitochondrial Genome Editing
Perhaps the most transformative breakthrough in mitigating the effects of mitochondrial heteroplasmy is the advent of direct mtDNA genome editing. Because traditional CRISPR-Cas9 systems rely on guide RNAs that cannot be effectively imported across the double-membrane into the mitochondrial matrix, mtDNA was considered completely "uneditable" for decades 6867.
This barrier was decisively broken with the engineering of DddA-derived cytosine base editors (DdCBEs). DdCBEs utilize split bacterial deaminase toxins fused to modular Transcription Activator-Like Effector (TALE) proteins, completely bypassing the need for guide RNA. Upon binding to adjacent sequences on the mtDNA, the split DddAtox halves reassemble and enable highly precise C•G-to-T•A conversions directly within the mitochondrial genome 56768.
In 2024 and 2025, researchers successfully utilized advanced configurations of these editors to permanently correct pathogenic mutations - such as the m.4291T>C variant linked to Gitelman-like syndrome - in patient-derived primary fibroblast colonies 68. Furthermore, engineered alpha-DdCBE variants have recently bypassed the traditional requirement for a thymine base immediately upstream of the target sequence (the 5'-T constraint). This biochemical advancement radically expands the editable range of the mitochondrial genome, allowing researchers to therapeutically target over 150 previously inaccessible heteroplasmic disease loci with high efficiency 368.
Organelle Transplantation
When the accumulation of deletions or heteroplasmy prevents direct editing, whole organelle replacement therapy offers a radical alternative. In 2026, researchers at the Chinese Academy of Sciences demonstrated the safe and highly efficient transplantation of healthy, exogenous mitochondria directly into target cells and living tissues.
By utilizing engineered red blood cell membrane vesicles as nanoscale "capsules," researchers successfully encapsulated functional mitochondria and delivered them into host cells. This specialized delivery vehicle bypassed cellular immune defenses and achieved an extraordinary 80% integration efficiency, allowing the new mitochondria to permanently fuse with the host's endogenous mitochondrial networks 69. In murine models of severe mitochondrial genetic disease, this capsule transplantation therapy successfully reversed widespread OXPHOS failure, rescued multi-organ degeneration, and significantly extended organismal lifespan by artificially diluting the total cellular heteroplasmic load 6970.
Conclusion
Mitochondrial heteroplasmy is not a static or incidental genetic condition, but rather a profoundly dynamic biological process operating at the critical nexus of genomics, cellular metabolism, and the passage of time. Driven initially by the inherently high mutation rate and unprotected architecture of the mitochondrial genome, somatic mutations undergo highly complex clonal expansions. These expansions are heavily influenced by random genetic drift in post-mitotic tissues, and, as recent longitudinal data confirms, distinct patterns of positive selection for deleterious variants in rapidly dividing cell lineages.
The aging complexity generated by mixed mitochondrial genomes is ultimately governed by the biochemical threshold effect. As silent mutations accumulate over decades, their detrimental effects remain effectively masked by the presence of compensatory wild-type mtDNA. It is only when tissue-specific bioenergetic tipping points are crossed that sudden, catastrophic declines in oxidative phosphorylation occur, triggering oxidative stress, metabolic reprogramming, and the rapid onset of systemic aging phenotypes.
With the maturation of ultra-sensitive duplex and single-cell sequencing methodologies, the true scale and functional consequences of this cellular heterogeneity are finally measurable. Consequently, the proactive mitigation of heteroplasmy - whether through the pharmacological stimulation of mitophagy, direct precise base-editing of the genome via DdCBEs, or whole organelle transplantation - represents one of the most promising, albeit complex, frontiers in the modern pursuit of extending human healthspan and treating age-related neurodegenerative diseases.