Microenvironmental failure in stem cell niche aging
Introduction: The Paradigm Shift in Regenerative Biology
The regenerative capacity of mammalian tissues relies entirely on the precise, orchestrated function of resident adult stem cells (ASCs). These multipotent progenitors are tasked with maintaining tissue homeostasis throughout an organism's lifespan and orchestrating complex repair mechanisms following injury or pathogenic insult 12. However, the foundational biological architecture governing these cells is not purely autonomous. The survival, quiescence, activation, and lineage commitment of ASCs are exquisitely dependent upon a highly specialized, localized microenvironment known as the stem cell niche 134. The niche comprises a dynamic, three-dimensional physical and biochemical network of supportive stromal and mesenchymal cells, extracellular matrix (ECM) scaffolds, metabolic and oxygen gradients, resident immune cells, and intricate vascular networks 45.
For decades, the biomedical consensus posited that somatic stem cell exhaustion was predominantly an intrinsic phenomenon, driven inexorably by cumulative DNA damage, telomere attrition, and chronological cellular fatigue 14. This intrinsic-centric paradigm was irrevocably disrupted by historical and modern iterations of heterochronic parabiosis. Building on the early shared-circulation experiments of Clive McCay in 1956, foundational studies - such as the landmark 2005 work by Conboy and colleagues - demonstrated that exposing aged, functionally dormant stem cells to a youthful systemic microenvironment could effectively restore their regenerative potential and reverse age-related functional decline 66.
This revelation precipitated a critical shift in aging research, leading to the current consensus: the microenvironment consistently fails before the stem cells do 17. Over chronological time, the stem cell niche undergoes an insidious, systemic degradation. This deterioration is characterized by extracellular matrix remodeling, progressive mechanical stiffening, localized metabolic shifts, and the accumulation of senescence-associated inflammatory factors 159. Aged niches fail to provide the requisite biochemical and biophysical cues necessary to maintain stem cell quiescence. Consequently, stem cells are subjected to aberrant activation signals, leading to cellular exhaustion, loss of stemness, and the eventual irreversible epigenetic and metabolic lockdown of the stem cell pool 78.
This report systematically examines the molecular and physical mechanisms driving the catastrophic collapse of the stem cell niche. It expands tissue coverage across highly regenerative organ systems - including the neural, cutaneous, muscular, hematopoietic, and intestinal compartments - and highlights the explosive growth of 2023 - 2026 single-cell RNA sequencing (scRNA-seq) and spatial transcriptomic data generated by geographically diverse research hubs spanning the Americas, Europe, Asia, and Africa 912101112. Furthermore, this analysis explores the pervasive controversies surrounding systemic rejuvenating factors, specifically Growth Differentiation Factor 11 (GDF11) 1314, and reviews the vanguard of clinical interventions targeting the microenvironment, including senolytics and in vivo partial epigenetic reprogramming 151916.
Mechanisms of Niche Failure: Extracellular Degradation and Mechanical Stiffening
The physical scaffold of the niche acts as a dynamic signaling interface rather than a mere structural support system. As chronological aging progresses, the biophysical properties of the niche undergo profound structural alterations that directly subvert stem cell function, transmitting pathological signals through mechanosensitive pathways 12.
Lysyl Oxidase (LOX) Cross-Linking and ECM Remodeling
In youthful tissues, the ECM is a finely tuned, viscoelastic network of collagens, laminins, and fibronectin that provides localized tethering for stem cells 1718. During aging, the delicate balance of ECM deposition and degradation is severely perturbed. Advanced glycation end-products (AGEs) steadily accumulate as persistent metabolic byproducts cross-link complex ECM molecules, restricting their flexibility and increasing tissue brittleness 2319. Concurrently, enzymatic cross-linking is pathologically upregulated. A primary driver of this is Lysyl oxidase (LOX), an enzyme responsible for the covalent cross-linking of collagens and elastin 119. While physiological LOX expression is critical for embryonic development and normal tendon mechanics, its dysregulation in aging niches drives pathological tissue fibrosis and extracellular stiffening 19. The regulatory networks controlling LOX are complex; for example, Transforming Growth Factor-beta 2 (TGF-$\beta$2) significantly upregulates LOX expression in mesenchymal stem cells, bridging fibrotic signaling with structural stiffening 19.
High-throughput quantitative proteomic studies reveal striking age-related remodeling of ECM components. In the skin and skeletal muscle, age-associated genomic stress (such as ultraviolet radiation exposure in the epidermis) induces a heterogeneous decrease in the expression of collagen type XVII $\alpha$1 chain (COL17A1) 1. The loss of COL17A1 leads to hemidesmosome fragility, causing the detachment of stem cells from their physical anchoring sites within the niche, which subsequently triggers their symmetric differentiation and depletion from the basal layer 118.
Mechanotransduction Failures and Piezo1 Hyperactivation
The downstream consequence of unchecked AGE accumulation and LOX-mediated cross-linking is an exponential increase in niche stiffness, a biophysical phenomenon that acts as a potent, non-cell-autonomous suppressor of stem cell activity 12319. Recent data highlight that stem cells process mechanical force through specialized mechanosensitive ion channels and cytoskeletal-nucleoskeletal mechanotransduction cascades 1720. In the central nervous system, atomic force microscopy and magnetic resonance elastography reveal that the subventricular zone (SVZ) and oligodendrocyte progenitor cell (OPC) niches stiffen drastically with age, correlating directly with the degradation of myelin and nervous system function 123.
To faithfully model this age-dependent mechanical transition in vitro, researchers have engineered stiffness-tunable hyaluronic acid-laminin hydrogels that match physiological hippocampal stiffness across different age groups 17. Culturing neural stem cells (NSCs) on these stiffened substrates demonstrates that mechanical forces alone are sufficient to accelerate the NSC aging phenotype, impairing their proliferation and neuronal differentiation 17. Hippocampal NSCs perceive this aberrant niche stiffness via the mechanosensitive ion channel Piezo1 1723. When the ECM stiffens, excessive Piezo1 activation triggers a mechanotransduction cascade that alters gene expression, upregulating collagen and integrin genes while drastically downregulating cell cycle-promoting genes 17. Disrupting Piezo1 signaling functionally rejuvenates aged NSCs, restoring their proliferative capacity and effectively blinding them to the stiffened microenvironment 1723.
Similarly, in bone marrow mesenchymal stem cells (BMSCs), increased matrix stiffness impairs the cytoskeletal-nucleoskeletal cascade 20. Stiff environments paradoxically reduce cellular traction forces in aged BMSCs, causing a decline in focal adhesion turnover and a pronounced reduction in total myosin IIa and Lamin A/C 20. This mechanical failure prevents the nuclear translocation of YAP/TAZ transcription factors. Consequently, the lack of nuclear YAP suppresses FOXO signaling pathways, downregulating antioxidant defense mechanisms and altering chromatin accessibility in regions critical for cellular senescence prevention 20.
Metabolic Shifts and the Senescence-Associated Secretory Phenotype (SASP)
Mechanical stress is intrinsically linked to localized metabolic failure. As niche support cells (such as pericytes, endothelial cells, and fibroblasts) age, they accumulate DNA damage, endure oxidative stress, and undergo cellular senescence 921. Rather than quietly undergoing apoptosis and clearing the tissue space, these cells linger in a state of irreversible cell cycle arrest, acting as "zombie cells" that secrete a toxic, pro-inflammatory cocktail known as the Senescence-Associated Secretory Phenotype (SASP) 212223.
The SASP is characterized by high concentrations of interleukin-6 (IL-6), interleukin-1$\beta$ (IL-1$\beta$), tumor necrosis factor-alpha (TNF-$\alpha$), matrix metalloproteinases (MMPs), and transforming growth factor-beta (TGF-$\beta$) 2223. Research from the University of Pompeu Fabra and the IRB Barcelona has demonstrated that the SASP effectively transforms a supportive stem cell niche into a hyper-inflamed, metabolically hostile zone that mirrors the inflammation associated with advanced systemic aging, even when experimentally induced in young tissues 21.
Niche inflammation forces stem cells out of their protective quiescent state. The continuous presence of SASP factors initiates an aberrant, localized hyper-proliferative response in surrounding stem cells, forcing them to divide and repair "phantom" injuries 59. This sustained metabolic overdrive depletes the stem cell pool, accelerating telomere attrition and forcing the remaining cells into deep, irreversible quiescence or apoptosis 59. In skeletal muscle, for instance, the fatty acid transporter CD36 is highly expressed in senescent niche cells, and its presence is strictly required for the active secretion of these pro-inflammatory SASP signals 21. Reducing the senescent cell load, or pharmacologically inhibiting CD36, dramatically attenuates inflammation and restores the regenerative functions of the stem cell niche 21.
Complex Signaling Crosstalk: Notch, Wnt, TGF-$\beta$, and BMP
The interaction between stem cells and their microenvironment relies heavily on highly conserved, finely tuned morphogen signaling pathways - predominantly Notch, Wnt, Bone Morphogenetic Protein (BMP), and TGF-$\beta$ 242531. During aging, the precise stoichiometric balance of these morphogens degrades, leading to catastrophic signaling misdirection.
Notch signaling normally requires direct cell-to-cell contact, utilizing ligands (Jagged 1/2, Delta-like 1/3/4) presented by a signal-sending niche cell to activate the Notch receptor on a signal-receiving stem cell 252633. Upon binding, a two-step proteolytic cleavage (mediated by ADAM10/TACE and the $\gamma$-secretase complex) releases the Notch Intracellular Domain (NICD), which translocates to the nucleus to drive the expression of target genes like Hes-1, promoting stemness, proliferation, and differentiation barriers 2533.
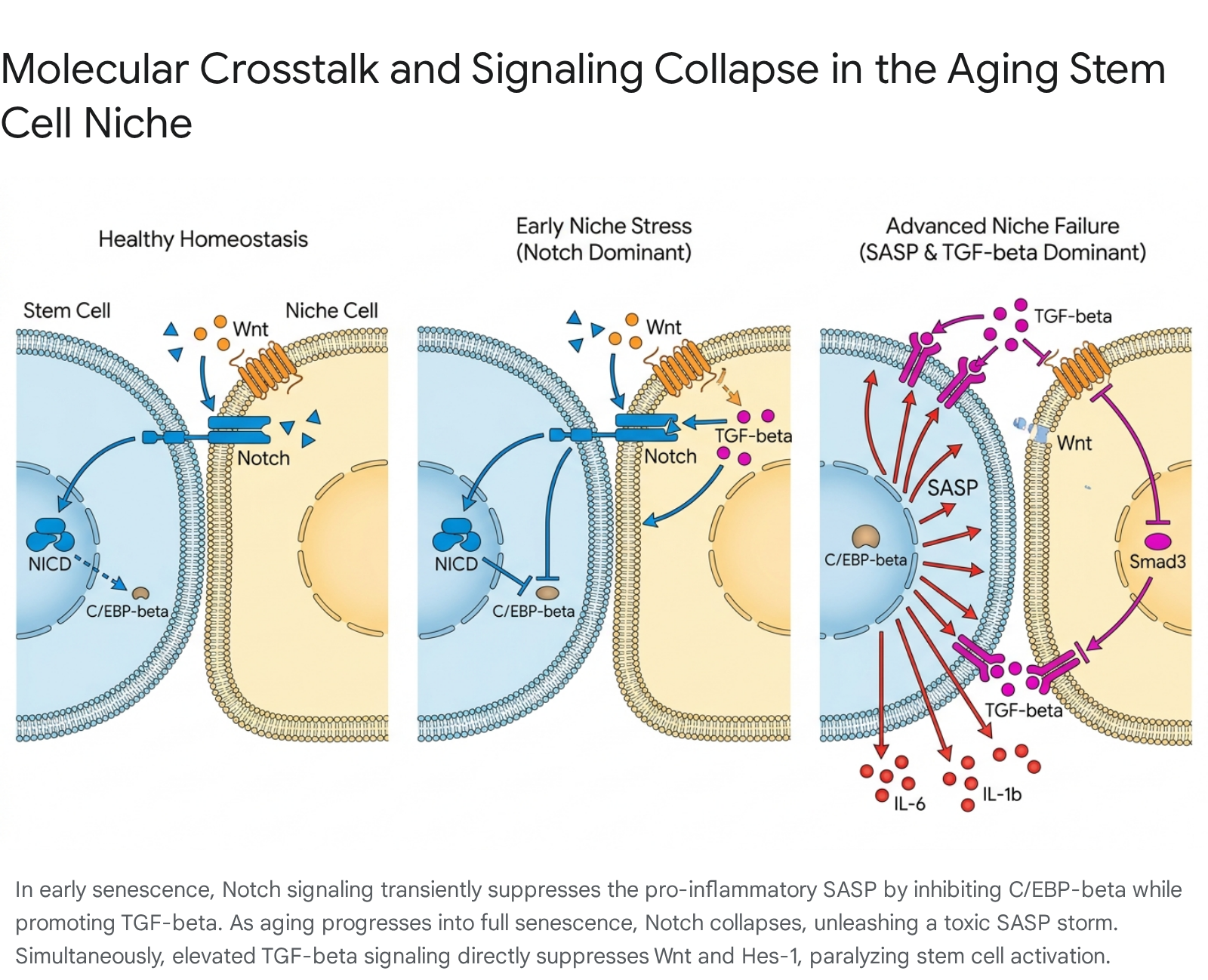
Recent evidence reveals a highly sophisticated, multi-state cross-talk between Notch, TGF-$\beta$, and the SASP during microenvironmental aging. In early, stress-induced senescence, transient Notch activity acts as a master regulator of the SASP 23. During this phase, the NICD drives a secretome rich in TGF-$\beta$ while actively suppressing canonical pro-inflammatory cytokines through the repression of the C/EBP$\beta$ transcription factor 23. Functionally, this Notch-high phase drives the juxtacrine, spatially restricted "lateral induction" of Notch signaling to neighboring cells 2325. However, as the niche reaches a state of "full senescence," Notch activity is precipitously downregulated. This downregulation releases the suppression of C/EBP$\beta$, triggering a massive local release of pro-inflammatory SASP factors, effectively switching the niche from a fibrotic state to a highly inflammatory one 23.
Concurrently, TGF-$\beta$ - which is abundant in the aged, fibrotic niche and heavily upregulated by the early SASP - directly interferes with the Notch signaling infrastructure. Smad3, an intracellular transducer of TGF-$\beta$, physically interacts with the NICD to synergistically alter the expression of the Notch target gene Hes-1 2427. Furthermore, elevated TGF-$\beta$ heavily downregulates Wnt signaling by directly suppressing the Wnt2b ligand, while simultaneously upregulating $\beta$-catenin 28. Because canonical Wnt/$\beta$-catenin signaling typically promotes stem cell activation and self-renewal across multiple compartments (such as the hair follicle and intestine), the TGF-$\beta$-driven suppression of Wnt effectively paralyzes the stem cells, locking them in a dormant, unresponsive state 18332930.
Organ-Wide Tissue-Specific Niche Exhaustion
The failure of the microenvironment is not a generalized pathology; it manifests distinctly depending on the unique cellular architecture, mechanical demands, and morphological layout of each organ system.
The Neural Stem Cell Niche
In the adult mammalian brain, neural stem cells (NSCs) are predominantly restricted to specialized niches in the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone (SGZ) of the dentate gyrus in the hippocampus 731. During healthy aging, the neural niche undergoes profound vascular dysfunction, leading to localized hypoxia and the disruption of critical circulating growth factors, such as Brain-Derived Neurotrophic Factor (BDNF) and Insulin-like Growth Factor 1 (IGF-1) 31. As previously noted, the physical stiffening of the neural ECM acts as a potent inhibitor of neurogenesis. Aged NSCs display prolonged quiescence and imbalanced differentiation, heavily favoring the generation of astrocytes over new neurons 731. Furthermore, the accumulation of senescent microglia within the niche generates a continuous SASP that mirrors the neuroinflammation observed in early-stage neurodegenerative conditions, significantly impairing learning, memory, and mood regulation 31.
Skin and Hair Follicle Niches
The hair follicle (HF) represents one of the most dynamic stem cell niches, undergoing continuous, lifelong cycles of growth (anagen), regression (catagen), and rest (telogen) 1832. Hair follicle stem cells (HFSCs) reside in a specialized, biochemically privileged compartment known as the bulge 3240. Aging within this niche results in the physical miniaturization of the hair shaft and the structural stiffening of the basement membrane 1830. Interestingly, recent intravital live-animal imaging has revealed a startling physical mechanism of stem cell exhaustion: as the surrounding niche structure atrophies, HFSCs physically escape from the stem cell compartment and migrate into the surrounding dermis, ultimately depleting the stem cell pool 33.
At the molecular level, hair cycling is governed by a competitive balance between BMP (which maintains HFSC quiescence during telogen) and Wnt (which drives activation during the transition to anagen) 1830. In aged HFSCs, this balance degrades. The activity of canonical Wnt/$\beta$-catenin signaling decreases, while the non-canonical, calcium-dependent Wnt pathway (driven by Wnt5a) is pathologically increased 30. This aberrant Wnt5a signaling increases the activity of Cdc42, resulting in cellular apolarity, structural disorganization, and the eventual exhaustion of the HFSC pool 30. Additionally, the miniaturization of the physical niche exerts mechanical compression on mechanosensitive Piezo channels, upregulating TNF-$\alpha$ and inducing widespread HFSC apoptosis 1830.
Skeletal Muscle Niche
Muscle stem cells (satellite cells) rely heavily on the elasticity of the surrounding basal lamina and the proximity of supportive capillary networks to function properly 121. In aging muscle, senescent cells emerge following standard micro-injuries and establish a hyper-inflamed environment 21. The fatty acid transporter CD36 is highly expressed in senescent muscle stem cells, driving the secretion of pro-inflammatory signals that blunt tissue repair at all stages of life 21. The reduction of the senescent cell burden - either genetically or pharmacologically - attenuates inflammation and remarkably improves muscle regeneration in both old and young models, emphasizing that niche inflammation is the primary blockade to regeneration rather than an intrinsic loss of satellite cell potential 21.
The Hematopoietic (Blood) Stem Cell Niche
The hematopoietic stem cell (HSC) niche within the bone marrow is highly complex, comprising endothelial cells, perivascular mesenchymal stromal cells (MSCs), osteoblasts, and complex sympathetic neural networks 3435. During aging, the bone marrow niche exhibits a phenomenon known as "mesenchymal drift," where supportive MSCs undergo an age-related differentiation bias 1634. Specifically, MSCs lose their osteogenic (bone-forming) potential and skew heavily toward adipogenic (fat-forming) differentiation, driven by an increase in the adipogenic marker PPAR$\gamma$ 534. As the marrow fills with adipocytes, the physical volume of the supportive endosteal niche collapses, displacing the HSCs further from the bone surface 4.
These newly formed adipocytes, alongside degenerating stromal cells, begin secreting inflammatory cytokines and reduce their production of essential retention factors like CXCL12 and Stem Cell Factor (SCF) 534. Concurrently, the aged marrow experiences a severe loss of sympathetic innervation. Proper sympathetic signaling via $\beta$2-adrenergic receptors is normally required to maintain the circadian clock (via the Per1 gene) and regulate HSC homing and egress 35. The loss of this regulation, combined with an inflamed niche, causes aged HSCs to exhibit a massive functional decline characterized by a distinct "myeloid bias" - a pathological overproduction of innate immune cells at the expense of adaptive lymphopoiesis, increasing the risk of both immunosenescence and myeloid leukemias 23436.
The Intestinal Stem Cell Niche
The intestinal epithelium is one of the fastest-regenerating tissues in the mammalian body, completely renewing every 3 to 5 days. This rapid turnover is driven by actively cycling Lgr5+ columnar base crypt (CBC) intestinal stem cells (ISCs) located at the base of the crypts of Lieberkühn 3373839. The ISC niche is maintained by a delicate interplay between epithelial Paneth cells and underlying mesenchymal stromal cells, including telocytes, trophocytes, and Foxl1+ winged-helix transcription factor-expressing cells 33140. These niche cells collaborate to maintain a crucial morphogen gradient: high Wnt and R-spondin at the crypt base to maintain stemness, and high BMP toward the villus to drive differentiation 32939. Essential BMP antagonists, such as Noggin and Gremlin 1 (Grem1), are secreted by pericryptal trophocytes to restrict BMP signaling within the stem cell zone 33139.
During aging, Paneth cell numbers are altered, and their secretion of antimicrobial peptides fluctuates. This perturbation heavily impacts the local microbiome and generates a state of chronic, low-grade inflammation termed "inflammaging" 384142. Recent studies from Kyoto University demonstrate that aging induces a profound imbalance between Interferon-gamma (IFN-$\gamma$) signaling (which becomes hyperactivated) and ERK/MAPK signaling (which is pathologically inactivated) 43. This shifting dynamic essentially forces the mammalian intestinal epithelium to prioritize the survival of the ISCs at the direct expense of differentiated cell maturation, contributing to systemic age-related metabolic shifts, reduced nutrient absorption, and compromised mucosal barrier integrity 3843.
Summary of Tissue-Specific Niche Decline
| Tissue System | Primary Stem Cell Target | Core Mechanism of Microenvironmental Failure | Niche Compositional Shift | Identifiers for Crosstalk |
|---|---|---|---|---|
| Neural | Neural Stem Cells (NSCs) | ECM mechanical stiffening; Piezo1 hyperactivation; Hypoxia. | Senescent microglia accumulation; Vascular dysfunction. | BDNF $\downarrow$, TGF-$\beta$ $\uparrow$ |
| Skin/Hair | Hair Follicle Stem Cells (HFSCs) | Basement membrane stiffening; Physical miniaturization of the shaft. | Physical escape of HFSCs into dermis; ECM atrophy. | BMP $\uparrow$, Canonical Wnt $\downarrow$ |
| Muscle | Satellite Cells | LOX-mediated collagen cross-linking; CD36+ driven senescence. | Fibrotic ECM deposition; Senescent cell accumulation. | SASP $\uparrow$, TGF-$\beta$ $\uparrow$ |
| Blood | Hematopoietic Stem Cells (HSCs) | Loss of sympathetic innervation; Reduced endosteal support volume. | MSCs differentiate into adipocytes; Inflammatory marrow. | CXCL12 $\downarrow$, IL-6 $\uparrow$ |
| Intestinal | Lgr5+ Intestinal Stem Cells (ISCs) | Inflammaging; Loss of morphogen gradient boundaries. | Paneth cell alterations; Trophocyte/Telocyte degradation. | Wnt $\downarrow$, IFN-$\gamma$ $\uparrow$ |
The Spatial Transcriptomic Revolution in Niche Research (2023 - 2026)
Understanding the immense complexity of the aging niche requires mapping the exact spatial relationships between stem cells and their neighboring support cells. Until recently, bulk RNA sequencing required tissue dissociation, effectively destroying spatial context and blending stem cells with their niche components into an indistinguishable average. Between 2023 and 2026, the advent of single-cell RNA sequencing (scRNA-seq) paired with high-resolution spatial transcriptomics (such as 10x Genomics Visium and Laser Capture Microdissection) has revolutionized niche mapping across the globe 944.
In the intestinal niche, spatial transcriptomics has generated unprecedented insights into cellular zonation along the crypt-villus axis. Utilizing combined scRNA-seq and spatial projection algorithms, researchers identified entirely novel niche components, including Lgr5+ telocytes located at the villus tip rather than the crypt base 9. Furthermore, spatial tracking during aging has revealed that "inflammaging" is not uniform. Single-cell mapping of the murine intestinal mucosa shows that different zones of the intestinal crypt acquire distinct chromatin accessibility shifts that propagate intrinsic sources of inflammation directly to the stem cells 945. Advanced computational models, such as the hierarchical Bayesian framework cSplotch, now allow researchers to integrate these massive, multi-modal datasets (encompassing over 400,000 single nuclei profiles), building topographical atlases that chart molecular aging gradients across entirely different organ systems 12.
Geographically diverse research cohorts have actively applied these technologies to uncover novel aging mechanisms in previously under-studied niches. In Asia, a 2023 study by Chinese researchers mapping the cranial suture mesenchyme - a unique niche for skeletal and mesenchymal stem cells - utilized scRNA-seq to pinpoint a specific senescent mesenchymal cell subset responsible for driving age-associated cranial inflammaging 1046. In parallel, global aging hubs in South Africa, working in collaboration with the Harvard Center for Population and Development Studies, have launched massive longitudinal cohorts integrating demographic aging patterns with high-resolution cellular markers. These initiatives aim to determine how endemic physiological stress, socioeconomic disparities, and the long-term effects of antiretroviral therapy (ART) scale-up accelerate biological niche deterioration in non-Western populations 12. Further structural support for this globalized approach comes from extensive funding initiatives within Latin America (spanning regenerative medicine priorities across Chile, Argentina, Brazil, Mexico, and Uruguay) and Europe (via Horizon Europe and organizations like the IRB Barcelona), which collectively emphasize that standardizing spatial transcriptomic datasets is paramount for developing universally applicable regenerative therapies 114748.
The GDF11 Controversy: Fact, Fiction, and Future Consensus
The investigation into systemic factors capable of rejuvenating the aged niche has been dominated by one of the most fiercely debated molecules in modern gerontology: Growth Differentiation Factor 11 (GDF11). A member of the TGF-$\beta$ superfamily, GDF11 rose to intense prominence following a series of heterochronic parabiosis studies in 2013 and 2014 by the Wagers and Lee laboratories. These foundational reports suggested that circulating GDF11 levels strictly decline with age, and that exogenous supplementation could profoundly reverse age-related cardiac hypertrophy, restore skeletal muscle regeneration, and revascularize the aging brain 61314.
Almost immediately, the biological community fractured. Multiple independent laboratories failed to reproduce the skeletal muscle rejuvenation effects, instead observing that high doses of GDF11 induced severe skeletal muscle fibrosis and actively accelerated functional decline in aged subjects 61349. The controversy was eventually traced back to profound methodological and biochemical challenges. The initial studies utilized SOMAmers and antibodies that lacked target specificity, heavily cross-reacting with Growth Differentiation Factor 8 (GDF8), more commonly known as Myostatin - a highly abundant and potent inhibitor of muscle growth 1314. GDF11 and GDF8 share 89% sequence similarity, differing by only 11 amino acids in their mature signaling domains, and are cleaved by different Tolloid proteases from prodomains that exhibit only 52% homology 131450. Despite this structural similarity, GDF11 is drastically more potent at inducing SMAD2 phosphorylation in target tissues 14. It was later revealed that the circulating pool was overwhelmingly dominated by GDF8, rendering the initial claims regarding the chronological decline of systemic GDF11 highly questionable 614.
As of 2024 - 2026, a more nuanced consensus has emerged, aided by refined analytical reagents and the involvement of targeted biopharmaceutical efforts (e.g., Elevian) 61314. The scientific community largely accepts that GDF11 exhibits potent, context-dependent regenerative capabilities, particularly in the central nervous system and the cardiovascular system. Recent data confirm that GDF11 acts as a pro-mnemonic and antidepressant agent, actively stimulating autophagy (via Beclin-1 upregulation) in aged hippocampi to clear senescent debris and restore neural stem cell pools 51. Furthermore, gene-therapy models utilizing controlled, inducible GDF11 expression in lung progenitors demonstrate significant attenuation of age-related pulmonary fibrosis following bleomycin injury, providing an "in situ factory" for targeted repair 60. Conversely, the role of GDF11 in skeletal muscle and hepatic fibrosis remains hotly contested, operating on a razor's edge where slight dosage variations flip the molecule from a regenerative agent into a pro-fibrotic trigger 64961. The current paradigm asserts that GDF11 is not a monolithic "magic bullet" anti-aging protein, but rather a highly potent, tissue-specific morphogen whose therapeutic window must be exquisitely controlled to effectively reprogram aged niches without inducing catastrophic fibrosis 131460.
Chronic Niche Dysfunction Driving Intrinsic Epigenetic and Metabolic Exhaustion
While aging begins as an extrinsic failure of the microenvironment, chronic exposure to a degraded niche ultimately forces stem cells across a biological event horizon, resulting in permanent intrinsic exhaustion 45. Stem cells possess high phenotypic plasticity, acting as environmental sensors. However, when subjected to unrelenting mechanical stress, localized hypoxia, and SASP exposure, the cellular machinery initiates survival mechanisms that paradoxically lock the cell into a dysfunctional state 79.
This irreversible lockdown is mediated primarily through profound epigenetic reprogramming. Dysregulated signaling from the niche alters the activity of DNA methyltransferases and histone deacetylases, leading to the permanent silencing of genes required for stem cell activation and the persistent opening of chromatin loci associated with senescence and inflammation 57. For instance, persistent oxidative stress induces hypermethylation of the promoter regions of essential DNA repair and cell-cycle genes, preventing the stem cell from responding to normal physiological cues 5. In highly proliferative environments like the intestine, Single-cell Assay for Transposase-Accessible Chromatin (scATAC-seq) data reveal that aged stem cells fundamentally alter their transcription factor networks, abandoning youthful regulatory architecture entirely 37.
Metabolic exhaustion runs parallel to epigenetic lockdown. A landmark 2025 study from the Icahn School of Medicine at Mount Sinai identified lysosomal hyperactivation and dysfunction as a central driver of stem cell aging 52. As hematopoietic stem cells reside in the aged, inflamed niche, their lysosomes - responsible for nutrient sensing and protein degradation - become hyperactive and highly acidic. This metabolic shift destroys the cell's ability to maintain proper catabolic and anabolic balance, flooding the cell with metabolic waste and driving systemic inflammation 52. Reversing this lysosomal hyperactivity successfully resets the stem cells to a youthful, healthy state, proving that metabolic exhaustion is a primary consequence of microenvironmental stress 52.
Furthermore, prolonged exposure to a failing niche disrupts global proteostasis. Proteomic evaluations of exosomes derived from quiescent hair follicle stem cells demonstrate a significant age-related depletion of critical translational machinery, specifically components of the 40S ribosomal subunit 8. This intrinsic decay is directly correlated to the upregulation of $p16^{INK4a}$, a potent cyclin-dependent kinase inhibitor that enforces permanent cell-cycle arrest 8. Consequently, even if these intrinsically exhausted stem cells are subsequently isolated and placed into a youthful, healthy niche environment, they often fail to engraft or fully regenerate tissue, acting as a testament to the devastating permanence of niche-induced epigenetic and metabolic memory 83637.
Therapeutic Vanguard: Senolytics and In Vivo Niche Reprogramming
Because extrinsic niche degradation acts as the primary catalyst for stem cell failure, modern regenerative medicine has pivoted sharply from simplistic, cell-replacement transplantation strategies toward systemic interventions designed to recalibrate and rejuvenate the microenvironment itself 2453.
Senolytics and the Suppression of Niche Inflammation
The most immediate, translational strategy for niche recovery is the targeted eradication of senescent support cells. Senolytics - pharmacological agents that selectively induce apoptosis in senescent cells - have shown profound efficacy in rapidly clearing the SASP and resolving localized tissue inflammation 1521. In a late 2024 human pilot study, a multi-agent therapeutic cocktail combining AM3 (a compound shown to enhance immune response), spermidine (a potent autophagy inducer), and hesperidin (a flavonoid antioxidant) yielded rapid reductions in circulating TNF-$\alpha$ and significantly rejuvenated immune cell profiles. This combination therapy effectively reset the inflammatory status of the hematopoietic niche, functionally reversing markers of immunoaging by an estimated ten years 15. In skeletal muscle, reducing the senescent cell burden directly mitigates CD36+ driven niche inflammation, actively restoring the regenerative capacity of resident satellite cells without requiring any direct manipulation of the stem cells themselves 21. Furthermore, pharmacological agents such as adrenergic beta-3 (ADR$\beta$3) agonists (e.g., BRL37344) are actively utilized to reset the sympathetic nervous regulation of the bone marrow niche, successfully blocking pathological myeloid expansion and functionally rejuvenating aged HSCs 36.
Localized In Vivo Epigenetic Reprogramming
The most disruptive, vanguard breakthrough in niche rejuvenation centers on in vivo partial epigenetic reprogramming 1954. Grounded in the foundational Nobel Prize-winning discovery of the Yamanaka factors (Oct4, Sox2, Klf4, and c-Myc, collectively OSKM), researchers determined that transient, carefully calibrated cyclic expression of these transcription factors can safely reset the epigenetic clock of somatic cells. Crucially, this partial reprogramming strips away accumulated epigenetic damage without pushing the cells entirely back into an undifferentiated, pluripotent state, thereby maintaining their essential somatic identity 166555.
Traditional in vitro reprogramming carries an extreme, inherent risk of oncogenesis and teratoma formation when translated to living subjects 19. However, precisely calibrated in vivo partial reprogramming circumvents this by limiting factor expression to a brief window (typically utilizing only OSK to avoid the heavily oncogenic c-Myc) 1654. By targeting the niche stromal and mesenchymal cells with these partial reprogramming vectors, the local microenvironment physically "forgets" its chronological damage, downregulating the SASP, repairing degraded ECM, and re-establishing youthful morphogen gradients 1655.
This technology is rapidly advancing toward clinical reality. At the 2025 Aging Research & Drug Discovery (ARDD) meeting in Copenhagen, industry leaders unveiled unprecedented progress. Life Biosciences announced that its partial epigenetic reprogramming technology (ER-100), designed to treat liver disease and optic neuropathies, is slated for human clinical trials in early 2026 16. Concurrently, Altos Labs detailed plans to utilize partial reprogramming to reverse "mesenchymal drift" in aged organs, rejuvenating the stromal microenvironment prior to transplantation 16. Furthermore, advanced commercial entities like Shift Bioscience are currently identifying single-gene alternatives (such as the proprietary SB000 vector) capable of bypassing the Yamanaka factors entirely, effectively decoupling cellular rejuvenation from dedifferentiation to ensure maximum in vivo safety 65. This technique aims to restore the microenvironment's capacity to naturally support and activate the endogenous stem cell pool, providing a scalable, localized intervention for systemic tissue aging 1654.
Conclusion
The biological aging of stem cells is not a self-contained, inevitable cellular tragedy, but rather a complex, systemic ecological collapse. The architectural deterioration of the stem cell niche - driven by LOX-mediated ECM stiffening, mechanotransduction failures through Piezo1 and YAP signaling, and the relentless inflammatory onslaught of the SASP - acts as the primary vanguard of tissue decay. Systemic miscommunication between essential pathways like Wnt, Notch, and TGF-$\beta$ transforms the localized microenvironment from a supportive cradle into an inhibitory prison, ultimately driving the stem cell pool into a state of irreversible epigenetic and metabolic exhaustion.
Driven by the unprecedented resolution of spatial transcriptomics and scRNA-seq, the global scientific community is now meticulously mapping these microenvironmental failures at the single-cell level. By resolving profound historical controversies surrounding systemic factors like GDF11, and pioneering highly targeted, revolutionary interventions such as senolytics and in vivo partial epigenetic reprogramming, a new therapeutic consensus has emerged. The future of regenerative medicine lies not merely in replacing the exhausted stem cell, but in resurrecting the niche that sustains it.