Mechanisms of vascular aging
The Vascular Theory of Aging
Vascular aging is increasingly recognized not merely as a localized deterioration of blood vessels, but as a primary, upstream determinant of systemic multiorgan decline. This paradigm, termed the vascular theory of aging, posits that age-related decreases in vascular function are the driving forces behind organismal aging at large 1234. Historically, the clinical understanding of vascular aging was confined to macrovascular pathologies, specifically arterial hypertension, arterial stiffness, and intimal atherosclerosis resulting in ischemic events 456. However, recent advances in vascular biology emphasize the profound impact of microvascular rarefaction, endothelial cellular senescence, and the disruption of organ-specific stem cell niches on overall longevity.
The cardiovascular system ages in a highly integrated manner. At the macrovascular level, structural alterations - such as the fragmentation of elastin, increased collagen deposition, and extracellular matrix (ECM) stiffening - alter the hemodynamic profile of the arterial tree, resulting in elevated pulse wave velocity and increased central pulse pressure 178. This stiffening subjects the microvasculature of highly perfused organs, such as the brain and kidneys, to pulsatile barotrauma. Concurrently, at the microvascular level, aging induces capillary rarefaction, compromised tissue oxygenation, and a localized failure of the endothelium to provide the necessary trophic support for parenchymal cell maintenance 4910.
Macrovascular Stiffening and Hemodynamic Alterations
The age-related remodeling of the large conduit arteries creates a hostile mechanical environment for the entire cardiovascular system. Arterial stiffening is driven by the degradation of elastic fibers and their gradual replacement by rigid collagen cross-links, a process exacerbated by advanced glycation end products (AGEs) 811. The loss of arterial compliance means the aorta can no longer dampen the pulsatile energy generated by the heart, transmitting high-pressure fluctuations directly into delicate microvascular beds 14.
This mechanical stress induces the expression of mechanosensitive ion channels and integrins in vascular smooth muscle cells (VSMCs). For example, recent studies utilizing VSMC-specific deletions of $\alpha$v integrins ($\alpha$vSMKO models) have demonstrated that the organization of focal adhesions directly contributes to the stiffness of the cells themselves, independent of the surrounding ECM 5. Under angiotensin II stimulation, $\alpha$v-deficient VSMCs exhibit increased cortical actin redistribution, driving a cellular stiffening response that precedes broader arterial wall stiffening 5. These mechanical shifts cause left ventricular hypertrophy, increased cardiac afterload, and eventually congestive heart failure 812.
Microvascular Rarefaction and VEGF Signaling Insufficiency
While the macrovasculature stiffens, the microvasculature undergoes progressive rarefaction - a structural reduction in capillary density. Reduced angiogenic potential is a hallmark of the aging vasculature, resulting from lower numbers of endothelial progenitor cells (EPCs), compromised cell survival, and a profound imbalance between pro- and anti-angiogenic factors 913.
Vascular Endothelial Growth Factor (VEGF) signaling insufficiency is a critical underlying mechanism of this age-related microvascular decline. Preclinical models demonstrate that a modest, compensatory increase in circulating VEGF is sufficient to preserve a youthful vascular homeostasis, alleviate multiple adverse age-related biological processes, and ameliorate a host of age-associated pathologies 23101314. Mice with genetic deletions of VEGF display rapid endothelial cell degeneration and death 1. The restoration of microvascular networks via VEGF augmentation highlights the bidirectional nature of tissue aging, wherein the vasculature not only passively responds to tissue damage but actively dictates the regenerative capacity of the surrounding tissue 213.
Angiocrine Signaling and Stem Cell Niche Deterioration
Beyond the mechanical delivery of oxygen and nutrients, vascular endothelial cells (ECs) function as an active paracrine and juxtacrine signaling hub. ECs secrete a diverse array of stimulatory and inhibitory growth factors, chemokines, cytokines, and ECM proteins collectively termed angiocrine signals 15161718. These signals are essential for tissue morphogenesis during embryogenesis and remain critical throughout adulthood for the maintenance of stem cell homeostasis, organ regeneration, and wound healing 151618. Aging severely disrupts the angiocrine output of the endothelium, precipitating systemic functional decline.
The Liver Sinusoidal Endothelial Niche
In the liver, liver sinusoidal endothelial cells (LSECs) form a highly specialized microvascular niche. Healthy LSECs are fenestrated and lack a continuous basement membrane, allowing for efficient metabolic exchange and the secretion of critical angiocrine factors, such as Wnt2 and Hepatocyte Growth Factor (HGF) 1819. These signals dictate hepatocyte proliferation and establish metabolic zonation across the hepatic lobule 18.
During aging, and in metabolic states such as metabolic dysfunction-associated steatohepatitis (MASH), LSECs undergo "capillarization." This phenotypic shift involves the loss of fenestrae and the formation of a rigid basement membrane 18. Defective, aging LSECs downregulate the c-kit receptor and upregulate adhesion molecules (VCAM-1) and chemokines (CCL2, CCL5), thereby promoting hepatic inflammation, fibrosis, and the progression of fatty liver disease 18. The impaired angiocrine signaling restricts hepatocyte regeneration, effectively halting the tissue's ability to recover from injury 1819.
Osteogenesis and Type H Vessels
In the skeletal system, osteogenesis is tightly coupled to the vascular supply. Specialized capillary subtypes, notably type H vessels - characterized by high expression of CD31 and Endomucin - coordinate bone remodeling dynamics by maintaining osteoprogenitor cells via angiocrine signaling 20.
With advancing age, the abundance of type H vessels declines significantly. This microvascular insufficiency, combined with a reduction in VEGF expression, limits the delivery of systemic nutrients and local angiocrine factors 20. The loss of endothelial-derived trophogens directly restricts osteoprogenitor cell activity, exacerbating uncoupled bone remodeling and accelerating age-related osteoporosis 1620. A parallel decline in osteoblast function further diminishes their ability to support lymphoid-biased hematopoietic stem cells, linking skeletal aging directly to immune system decline 16.
The Bone Marrow Hematopoietic Niche
The impact of angiocrine dysfunction is most profoundly observed within the bone marrow hematopoietic stem cell (HSC) niche. Bone marrow endothelial cells (BMECs) form a critical supportive microenvironment for HSCs. In aged murine models, BMECs exhibit increased vascular permeability, localized hypoxia, and elevated production of reactive oxygen species (ROS), alongside a marked reduction in the expression of essential HSC maintenance factors such as Stem Cell Factor (SCF) and CXC motif chemokine ligand 12 (CXCL12) 16.
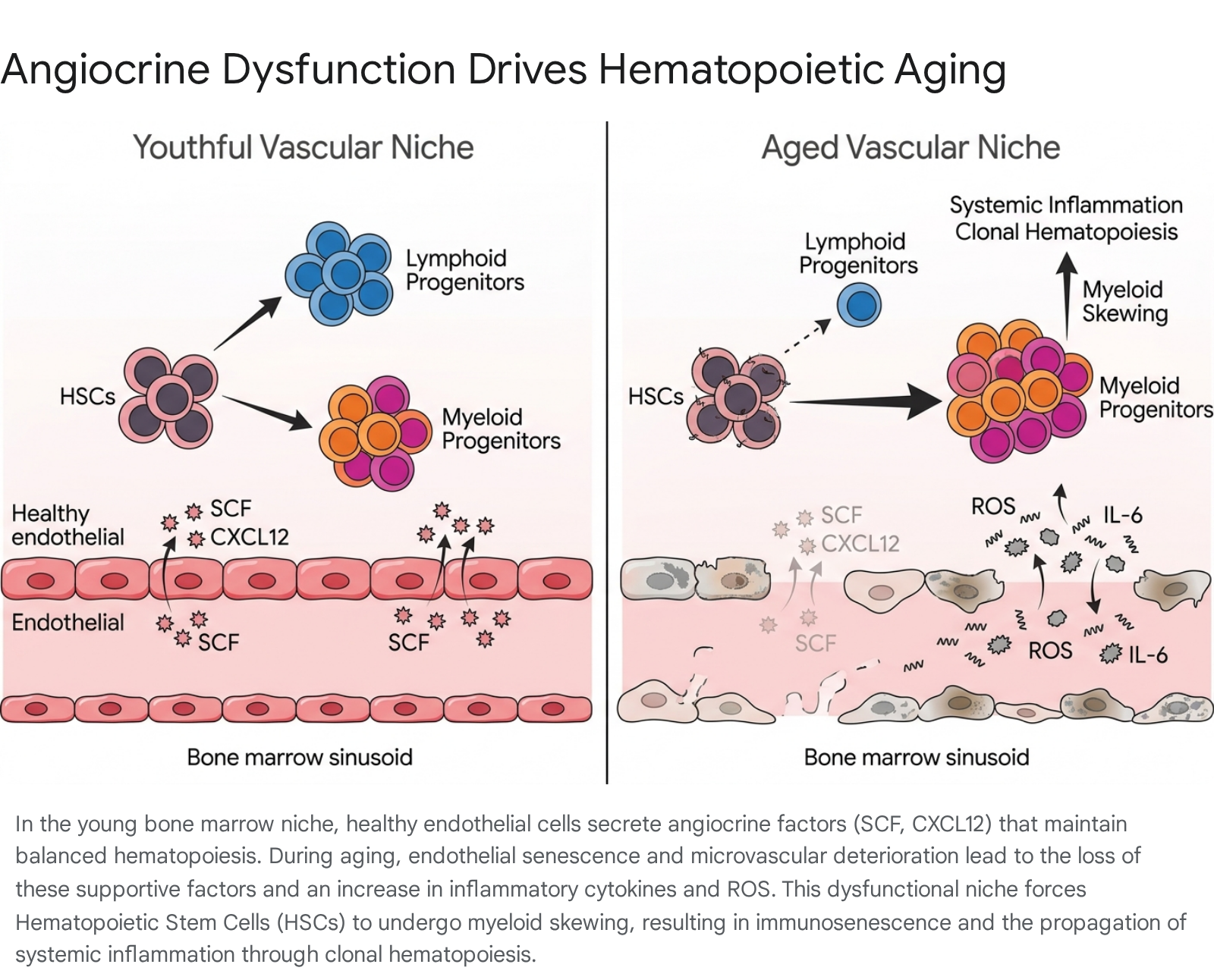
This deterioration of the vascular niche directly influences the intrinsic aging of HSCs. The disruption of angiocrine signaling skews HSC differentiation away from lymphoid lineages and toward myeloid lineages 16. This age-associated myeloid bias contributes heavily to immunosenescence, rendering older adults more susceptible to infections and reducing the adaptive immune system's efficacy 1621.
Furthermore, this niche dysfunction provides a selective advantage to HSCs harboring specific age-associated somatic mutations (e.g., TET2, DNMT3A, ASXL1). The expansion of these mutant hematopoietic clones, a condition known as Clonal Hematopoiesis of Indeterminate Potential (CHIP), acts as a systemic amplifier of age-related disease 16. Mutant macrophages derived from CHIP clones infiltrate the arterial wall and systemic organs, driving aberrant systemic inflammation and severely accelerating vascular dysfunction, atherosclerosis, and heart failure risk 16. The experimental infusion of aged ECs into young animals has been shown to successfully replicate these detrimental effects on hematopoietic recovery and lineage balance, providing direct evidence that endothelial aging is a causal driver of hematopoietic decline 16.
Medial Calcification and Vascular Smooth Muscle Cell Plasticity
While intimal calcification is primarily driven by lipid deposition, macrophage infiltration, and localized inflammation (atherosclerosis), medial vascular calcification is a distinct age-related pathology characterized by the deposition of calcium phosphate crystals within the tunica media of arteries 2223. Medial calcification is highly prevalent in aging populations, as well as in patients with chronic kidney disease (CKD) and diabetes mellitus, leading to severe arterial stiffening, left ventricular hypertrophy, and eventual cardiovascular mortality 82224.
The Phenotypic Switch
Historically, cardiovascular calcification was viewed as a passive, degenerative precipitation of calcium. Advanced lineage tracing and single-cell transcriptomics have redefined it as a highly active, cell-mediated process governed by the phenotypic plasticity of vascular smooth muscle cells (VSMCs) 122526.
In a healthy adult vessel, VSMCs maintain a physiological contractile phenotype characterized by a low proliferation rate and the robust expression of specific contractile markers, such as alpha-smooth muscle actin ($\alpha$-SMA) and SM22$\alpha$ 232627. This phenotype provides mechanical strength and regulates vascular tone. However, differentiated VSMCs retain remarkable plasticity. In response to aging, oxidative stress, hyperglycemia, or hyperphosphatemia, VSMCs downregulate their contractile markers and undergo phenotypic switching 242526.
While VSMCs can adopt macrophage-like, myofibroblast-like, or synthetic phenotypes, the transition to an osteochondrogenic (bone-like or cartilage-like) state is the absolute prerequisite for medial calcification 82226. These transdifferentiated VSMCs actively produce a local pro-calcifying environment. They secrete matrix vesicles - exosome-like structures loaded with calcium, phosphate, and specific microRNAs - that serve as nidus sites for the precipitation and growth of hydroxyapatite crystals in the extracellular matrix 8122225.
| Phenotypic State | Primary Function | Key Molecular Markers | Secretory Profile | Vascular Pathophysiology |
|---|---|---|---|---|
| Contractile VSMC | Regulate vascular tone and diameter | $\alpha$-SMA, SM22$\alpha$, Calponin | Elastin, collagen (structural matrix) | Normal physiological state |
| Synthetic VSMC | Tissue repair and proliferation | Decreased $\alpha$-SMA, increased PDGF receptors | High ECM production, proteases | Intimal hyperplasia, vascular remodeling |
| Macrophage-like VSMC | Lipid scavenging and inflammation | CD68, Mac-2, ABCA1 | IL-1$\beta$, TNF-$\alpha$, MCP-1 | Atherosclerotic plaque formation (foam cells) |
| Osteochondrogenic VSMC | Active matrix mineralization | RUNX2, MSX2, ALPL, BMP2 | Calcifying matrix vesicles, reduced MGP | Medial arterial calcification, arterial stiffness |
Intracellular Drivers of Transdifferentiation
The osteochondrogenic transdifferentiation of VSMCs mirrors physiological endochondral bone formation. It is driven by the activation of complex intracellular signaling cascades, most notably the Wingless-related integration site (Wnt)/$\beta$-catenin pathway and Bone Morphogenetic Protein (BMP) signaling 222528.
Extracellular phosphate functions not merely as a mineral substrate, but as a potent signaling molecule 22. Under high phosphate conditions (common in CKD and localized aging microenvironments), the Wnt cascade is activated. This leads to the stabilization and nuclear translocation of $\beta$-catenin, which upregulates the transcription factor RUNX2 (Runt-related transcription factor 2) 2528. RUNX2 is the master regulatory gene for osteoblast differentiation. Its expression is normally undetectable in healthy blood vessels but is highly upregulated in calcified arteries, where it induces the expression of bone-related proteins like osteocalcin, bone sialoprotein, and tissue non-specific alkaline phosphatase (TNAP/ALPL) 122728. Loss-of-function studies confirm that smooth muscle cell-specific RUNX2 suppression dramatically inhibits medial calcification progression in both atherosclerosis-susceptible and CKD murine models 28.
BMP-2 also plays a critical role by activating Smad1/5 signaling, further promoting the osteogenic program in VSMCs 222324. This pathway can be triggered by excessive hyperinsulinemia, where the transcription factor MSX2 works in concert with BMP-2 to drive calcification in models of insulin resistance 24. Conversely, healthy vessels actively suppress this process via potent endogenous mineralization inhibitors, such as Matrix Gla Protein (MGP) and Fetuin-A 82327. MGP binds directly to BMP-2 to inhibit its activity; thus, the loss of MGP expression during VSMC dedifferentiation results in unopposed BMP-2 signaling, accelerating the calcification cascade 823. Adiponectin has also been identified as a critical inhibitor of this transformation, blocking the transition of VSMCs via the inhibition of the mTOR and JAK2-STAT3 pathways 24.
Mineral Stress and Nuclear Lamina Dysfunction
Recent molecular profiling has identified epigenetic disruption as an early, upstream driver of this phenotypic switch. In 2025, a landmark study from the Shanahan group demonstrated that mineral stress directly induces profound changes in the nuclear envelope, specifically through the accumulation of prelamin A, a hallmark of nuclear lamina dysfunction previously associated with progeria syndromes (e.g., Hutchinson-Gilford progeria syndrome) 29.
The accumulation of uncleavable prelamin A in human VSMCs disrupts chromatin architecture by reducing histone methyltransferase activity while simultaneously increasing the activity of histone demethylases 29. This coordinated enzymatic shift depletes the critical repressive histone marks H3K9me3 (trimethylated histone 3 lysine 9) and H3K27me3 (trimethylated histone 3 lysine 27), leading to widespread heterochromatin loss 29. Crucially, this epigenetic remodeling, which triggers VSMC senescence and the subsequent upregulation of osteogenic genes, occurs temporally prior to the onset of actual calcification 29. This establishes epigenetic heterochromatin loss as an early harbinger of vascular inflammaging and a potential target for intercepting the pathology before irreversible mineral deposition occurs.
Oxidative Stress and Redox Imbalance
Oxidative stress is intimately linked to the induction of vascular senescence and calcification. The "Oxi-Inflamm-Aging" theory posits that the continuous accumulation of reactive oxygen species (ROS) disrupts the metabolic microenvironment, damaging DNA and triggering the senescence cascade 730.
Recent transcriptomic analyses have highlighted the dysregulation of the glutathione (GSH) pathway - the major cellular antioxidant system - during vascular aging. Specifically, the expression of the glutamate-cysteine ligase modifier subunit (GCLm) is significantly elevated in senescent, calcifying VSMCs and in unstable human atherosclerotic plaques 27. In VSMCs exposed to high-phosphate environments, the GSH:GSSG ratio (reduced to oxidized glutathione) plummets, indicating a severe redox imbalance 27. Experimental forced overexpression of GCLm in murine VSMC models significantly accelerates calcification and the loss of the contractile phenotype, implicating precise nodes of glutathione metabolism as both markers and drivers of the age-related calcific phenotype 27.
Cellular Senescence and the Aging Vasculature
Cellular senescence is a fundamental hallmark of aging characterized by a permanent, stable state of cell cycle arrest induced by intrinsic or extrinsic stressors, including telomere attrition, DNA damage, oxidative stress, and mechanical stiffening 41430. In the context of vascular aging, both ECs and VSMCs are highly susceptible to senescence, leading to a progressive accumulation of senescent cells within the arterial wall 61030.
The Senescence-Associated Secretory Phenotype
Senescent vascular cells are not metabolically inert; they undergo widespread transcriptomic changes and develop a Senescence-Associated Secretory Phenotype (SASP) 43031. The SASP is a complex, pro-inflammatory secretome consisting of cytokines (e.g., IL-6, IL-1$\beta$, TNF-$\alpha$), chemokines (MCP-1), growth factors (TGF-$\beta$), and matrix metalloproteinases (MMPs) 62030.
This localized secretory profile initiates a vicious cycle of vascular degradation. In the endothelium, SASP factors suppress the expression and activity of endothelial nitric oxide synthase (eNOS) via nuclear factor-$\kappa$B (NF-$\kappa$B) signaling 3032. The resulting decline in nitric oxide bioavailability cripples endothelium-dependent vasodilation, promoting hypertension 30. Concurrently, within the medial layer, SASP-derived TGF-$\beta$ and MMPs drive pathological ECM remodeling, collagen cross-linking, and further VSMC stiffening, structurally amplifying the aging process 46. The SASP essentially acts as an autocrine and paracrine amplifier, spreading senescence to adjacent healthy cells and recruiting immune cells that chronically sustain vascular inflammation 63033.
Endothelial Senescence and Neurovascular Decline
The implications of endothelial senescence are particularly severe in the central nervous system. Brain endothelial cells exhibit heightened sensitivity to aging-induced senescence, transitioning into an arrested state at a greater rate and earlier than other brain cell types, often observable by middle age 2. The loss of genomic stability, specifically through deficiencies in excision repair cross complementation group 1 (ERCC1), has been shown to strongly promote this cellular aging in ECs 6.
This microvascular senescence drives profound neurovascular dysfunction. It impairs neurovascular coupling (NVC) - the ability of blood vessels to dynamically dilate in response to localized neuronal activity - while inducing microvascular rarefaction and the breakdown of the blood-brain barrier (BBB) 26. These cerebrovascular alterations limit the speed and amplitude of hemodynamic responses to neuronal demand, playing a significant causal role in the development of vascular cognitive impairment (VCI) and broader neurodegenerative diseases 26. Experimental heterochronic parabiosis studies confirm that exposing young mice to an aged humoral milieu accelerates the acquisition of these neurovascular aging traits, highlighting the systemic nature of the decline 2.
Epigenetic Clocks and Biological Age Profiling
Aging is characterized by highly reproducible alterations in DNA methylation patterns. These epigenetic changes are sufficiently predictable that they have been harnessed to create "epigenetic clocks," which utilize machine-learning algorithms to calculate a biological age that often differs from a patient's chronological age based on the methylation status of specific cytosine-guanine (CpG) dinucleotides 34353637.
Clock Generations and Vascular Vulnerability
The technology underlying epigenetic clocks has evolved rapidly. First-generation clocks, such as the Horvath clock (a multi-tissue predictor based on 353 CpG islands) and the Hannum clock (blood-specific based on 71 CpGs), were trained simply to predict chronological age by comparing methylation signatures of young and old individuals 3536.
Second and third-generation clocks were specifically calibrated to capture physiological deterioration and mortality risk. Second-generation models like PhenoAge and GrimAge model health phenotypes and predict the risk of age-related clinical outcomes 3536. Third-generation models, notably DunedinPACE, measure the dynamic pace of multi-organ system deterioration over time 3436.
| Clock Generation | Example Models | Primary Training Target | Clinical Application Focus |
|---|---|---|---|
| First Generation | Horvath Clock, Hannum Clock | Chronological Age | Basic biological aging assessment; forensic age estimation |
| Second Generation | PhenoAge, GrimAge | Mortality Risk & Healthspan | Predicting cardiovascular events, lifespan, and comorbidity risk |
| Third Generation | DunedinPACE | Rate of Physiological Decline | Measuring the speed of aging; evaluating gerotherapeutic interventions |
| Targeted Clocks | Cell-Type Resolution Clocks | Specific Cell Aging (e.g., glia, LSECs) | Isolating accelerated aging in specific tissues (e.g., brain in Alzheimer's) |
Vascular-specific epigenetic signatures have demonstrated profound clinical utility in predicting cardiovascular outcomes independently of chronological age. Large-scale population analyses, such as the Multi-Ethnic Study of Atherosclerosis (MESA), show that accelerated epigenetic aging (e.g., a higher GrimAge relative to chronological age) strongly correlates with an increased subclinical atherosclerotic burden and incident cardiovascular events 3238. In fully adjusted models analyzing 1,264 participants, GrimAge acceleration was independently associated with a higher risk of composite cardiovascular disease (HR, 1.05), stroke (HR, 1.08), and heart failure with mildly reduced ejection fraction (HR, 1.31) 38. Among patients with detectable Coronary Artery Calcium (CAC > 0), epigenetic acceleration remained a significant predictor of myocardial infarction (HR, 1.09) 38. Furthermore, longitudinal studies published in late 2025 confirmed that modifiable cardiovascular risk factors - such as poor glycemic control, high body mass index (BMI), and smoking - directly accelerate these methylation-based aging trajectories, while physical activity and diet slow the pace of aging 39.
Tissue Specificity and Future Directions
While these models are highly predictive, standardization remains a clinical challenge. Most commercial epigenetic clocks are trained on blood samples, which consist entirely of leukocytes with well-characterized methylation profiles 3536. However, commercial testing often relies on saliva samples, which are mixtures of immune cells (~65%) and buccal epithelial cells (~35%). Applying blood-trained algorithms to saliva without sophisticated correction introduces substantial error margins, as the methylation signatures of the different cell types vary fundamentally 36.
Recent advancements aim to bypass bulk tissue limitations by advancing to cell-type resolution. By developing advanced computer models, researchers can now isolate and quantify the epigenetic age of precise cellular populations from complex tissues 37. For instance, cell-type specific assessments have revealed that brain glia age at a drastically accelerated rate in patients with Alzheimer's disease compared to other brain cells, and that specific liver cells exhibit accelerated epigenetic aging in fatty liver disease 37.
Epidemiological Divergence in Vascular Aging
While the molecular mechanisms of vascular aging are universal, their clinical manifestation exhibits significant epidemiological variance. Chronological age remains the dominant risk factor for cardiovascular disease globally, yet longitudinal cohort studies reveal stark disparities in the onset and velocity of vascular aging across different racial and ethnic groups 404142.
Accelerated Risk in South Asian Populations
Populations of South Asian descent (originating from countries such as Bangladesh, India, Pakistan, Nepal, and Sri Lanka) exhibit a highly accelerated phenotype of vascular aging 414243. Despite aggregated statistics often portraying Asian Americans as having lower ASCVD risk (largely due to lower rates in East Asian cohorts), disaggregated data reveals that South Asians suffer from disproportionately high rates of ASCVD and experience myocardial infarctions at a median age of 53, a full decade earlier than populations of European or Chinese descent 414344.
Comparative longitudinal analyses combining data from the MASALA (Mediators of Atherosclerosis in South Asians Living in America) and MESA (Multi-Ethnic Study of Atherosclerosis) cohorts provide high-resolution insights into this divergence 414245. The data indicates that South Asian adults develop critical antecedents of vascular aging - specifically insulin resistance, endothelial dysfunction, and hypertension - significantly earlier in life 414245. Echocardiographic studies assessing the E/e′ ratio (a surrogate for left ventricular filling pressure and diastolic dysfunction) from the E-ECHOES study demonstrate that South Asians undergo significantly accelerated, age-dependent cardiac and vascular stiffening compared to African Caribbean and White cohorts, and that this premature aging is an independent predictor of mortality 46.
| Cardiovascular Risk Factor Prevalence (at Age 45) | South Asian Men | White Men | Black Men | Hispanic Men | Chinese Men |
|---|---|---|---|---|---|
| Prediabetes | 30.7% | 3.9% | 10.4% | 10.5% | 12.6% |
| Hypertension | 25.0% | 18.0% | Similar/Higher | 10.0% | 6.0% |
| High Cholesterol/Triglycerides | 78.0% | - | 61.0% | - | - |
Data compiled from the combined longitudinal analysis of the MASALA and MESA cohort studies 4345. Note: South Asian women exhibit parallel accelerated risk, with prediabetes rates at age 45 (17.6%) significantly higher than White (5.7%), Black (9.0%), Chinese (8.2%), and Hispanic (5.1%) women 45.
The Behavioral Paradox
The accelerated vascular aging seen in South Asians presents a complex clinical paradox. Despite exhibiting higher rates of severe microvascular and macrovascular risk factors by age 45, South Asian cohorts in the MASALA study consistently report behavioral metrics that typically protect against vascular aging. Specifically, they report significantly higher diet quality scores, lower alcohol consumption, and comparable or superior leisure-time exercise habits relative to other ethnic groups 41424345. (While environmental and behavioral toxins generally accelerate vascular aging - for instance, the heavy concentration of CB1 receptors on the developing and adult cardiovascular system makes the vasculature highly susceptible to arteriopathic damage from cannabis use - such behaviors do not account for the divergence seen in this demographic 49).
This mismatch between ostensibly healthier lifestyle behaviors and severe clinical risk suggests that traditional framings of ASCVD are insufficient to explain the etiology in the South Asian population 43. Researchers hypothesize that the divergence is driven by a combination of unique genetic predispositions (such as variations in the APOA1 and APOA2 genes regulating lipid metabolism), altered body fat distribution (higher visceral adiposity at lower or normal BMIs), and potential early-life environmental stressors that epigenetically program the vascular system for accelerated metabolic decline 4344. By age 55, South Asian adults demonstrate the highest estimated hazard probability of clinical diabetes among all studied racial and ethnic groups, necessitating earlier and more aggressive screening protocols 424345.
Emerging Pharmacological Interventions
The recognition of vascular aging as a modifiable biological process, rather than an inevitable deterioration, has catalyzed the development of targeted gerotherapeutics. These interventions aim to delay, halt, or reverse the underlying hallmarks of aging within the vasculature by targeting cellular senescence, systemic inflammation, and nutrient sensing 33395047.
Senolytics and Immune-Based Senolysis
Senolytics are a class of drugs designed to selectively induce apoptosis in senescent cells by targeting their unique pro-survival pathways (e.g., BCL-2 family proteins). The combination of Dasatinib (a tyrosine kinase inhibitor) and Quercetin (a flavonoid) - collectively known as D+Q - was the first generation of senolytics to demonstrate efficacy 3350. In early human pilot studies, D+Q significantly reduced the burden of senescent cells in adipose tissue and lowered circulating SASP factors in patients with diabetic kidney disease, providing crucial proof-of-biology for systemic senolysis 5048.
Navitoclax (ABT-263), a potent BCL-xL inhibitor, has shown high efficacy in clearing senescent vascular and hematopoietic stem cells in preclinical models 313350. However, systemic BCL-xL inhibition faces clinical hurdles due to on-target dose-limiting toxicities, specifically severe thrombocytopenia, as platelets rely heavily on BCL-xL for survival 50. Consequently, the senolytics field is pivoting toward localized delivery to mitigate systemic toxicity. A prominent example is UBX1325, a senolytic BCL-xL inhibitor administered directly into the eye, which has shown positive efficacy readouts in Phase 2 trials for diabetic macular edema by locally clearing senescent vascular cells in the retina 50.
Next-generation strategies are actively exploring immune-based senolysis. These approaches apply immuno-oncology principles to overcome the immune evasion tactics of senescent cells. Emerging strategies include blocking immunosuppressive ligands (e.g., GD3 ganglioside) and engineering chimeric antigen receptor (CAR) T cells to specifically target senescence surface markers like the urokinase-type plasminogen activator receptor (uPAR) 33. Concurrently, senomorphics (or senostatics) aim to suppress the SASP without killing the cell. Inhibitors of the NF-$\kappa$B pathway and targeted neutralizing antibodies are under investigation for their ability to blunt the pro-inflammatory impact of senescent ECs and VSMCs 33.
NLRP3 Inflammasome Inhibition
The NLRP3 inflammasome is an intracellular sensor that detects danger signals and environmental insults, triggering a powerful pro-inflammatory cascade via the activation of Caspase-1 and the release of IL-1$\beta$ and IL-18 49. The chronic, sterile activation of NLRP3 is a fundamental driver of age-related vascular inflammation (inflammaging), atherosclerosis, and metabolic dysfunction 54.
Therapeutic targeting of NLRP3 has yielded highly promising clinical data. BGE-102, a structurally novel, orally available, and brain-penetrant small-molecule NLRP3 inhibitor developed by BioAge Labs, has demonstrated profound anti-inflammatory efficacy in early-phase human trials 5450. In a Phase 1 multiple ascending dose (MAD) trial in patients with obesity and elevated baseline inflammation, a 120 mg daily dose of BGE-102 achieved an 86% median reduction in high-sensitivity C-reactive protein (hsCRP) by day 14 5851. Remarkably, 93% of participants reached hsCRP levels below 2 mg/L, the clinically recognized threshold for reduced cardiovascular risk 51. The drug also achieved a 58% reduction in IL-6, a 30% reduction in fibrinogen, and near-complete (93%) suppression of IL-1$\beta$ 51. A newly evaluated 60 mg once-daily cohort demonstrated similar best-in-class reductions 58.
Supported by this Phase 1 data, BGE-102 is advancing into a Phase 2 dose-ranging proof-of-concept trial targeting cardiovascular risk, expected to initiate in mid-2026 with initial data by year-end 54585253. Concurrently, based on preclinical models demonstrating that oral BGE-102 prevents retinal vascular leakage and lipofuscin accumulation, the compound is entering a Phase 1b/2a trial for diabetic macular edema (DME) in mid-2026, positioning it as a potential "pipeline in a pill" capable of addressing NLRP3-driven inflammation across cardiovascular, ocular, and central nervous systems 5853.
Modulating Nutrient Sensing and Hypoxia
The mechanistic target of rapamycin (mTOR) is a highly conserved nutrient-sensing kinase that regulates cellular metabolism, growth, and survival. Hyperactivation of mTOR signaling during aging inhibits autophagy and accelerates cellular senescence 1454. Rapamycin, a specific mTOR inhibitor conventionally used as an immunosuppressant at high doses, acts as a potent geroprotector at lower, intermittent doses 3354.
Inhibition of the mTOR pathway mimics the systemic benefits of caloric restriction. In vascular models, rapamycin suppresses the SASP, enhances autophagic clearance of damaged organelles (mitophagy), and delays endothelial dysfunction 144854. Furthermore, clinical trials in older adults have demonstrated that low-dose mTOR inhibition can significantly improve immune function, upregulate antiviral responses to vaccination, and reduce markers of cellular senescence (such as p16INK4A) in topical applications 4854. The ongoing Targeting Aging with Metformin (TAME) trial and various human studies of rapamycin analogs (rapalogs) are actively evaluating the capacity of nutrient-sensing modulation to delay the onset of cardiovascular and age-related functional decline 55.
Other biological pathways targeted to reverse vascular aging include hypoxia signaling and APJ receptor agonism. BGE-117, a small-molecule inhibitor of Hypoxia-Inducible Factor Prolyl Hydroxylase (HIF-PH), activates HIF signaling to simulate a cellular response to low oxygen. This activation improves oxygen delivery, boosts erythropoiesis, and promotes vascular remodeling and angiogenesis . By boosting biological processes controlled by HIF target genes, BGE-117 has been evaluated in Phase 2a clinical trials for unexplained anemia of aging (UAA), attempting to counteract the progressive decline in microvascular oxygen transport . Furthermore, compounds targeting immune aging directly, such as BGE-175 (an inhibitor of the PGD2-DP1 pathway), have shown the ability to reverse age-related declines in dendritic cell migration, restoring the immune system to a more youthful state capable of surviving lethal viral challenges in aged models 56.
Metabolic Combinations and Incretin Therapies
Metabolic dysfunction directly drives vascular aging. The advent of highly effective incretin therapies, such as the GLP-1/GIP receptor agonist tirzepatide, has revolutionized the management of obesity and diabetes. In Phase 3 SURMOUNT trials, tirzepatide resulted in profound weight loss (up to 20.9% reduction over 72 weeks) and significant improvements in cardiovascular risk profiles, even among patients taking concomitant weight-inducing medications 5758. The clinical pipeline is rapidly expanding to include oral incretins (e.g., orforglipron) and novel combinations to maximize metabolic and vascular benefits 5168.
Attempts to target metabolic and vascular aging simultaneously via combination therapies highlight the field's rapid innovation, though not without safety challenges. Azelaprag (BGE-105), an oral agonist of the apelin receptor (APJ), demonstrated synergistic, additive weight loss and restored body composition when combined with incretin therapies like tirzepatide in preclinical models, theoretically preserving muscle mass while enhancing fat loss 50. However, the Phase II STRIDES trial evaluating azelaprag in combination with tirzepatide was halted in late 2024 after 11 participants exhibited asymptomatic elevations in liver transaminases, illustrating the complex safety hurdles inherent in modulating systemic metabolic and vascular receptors 59. Conversely, combinations such as Eli Lilly and Regeneron's tirzepatide plus mibavademab (an experimental antibody) have successfully completed Phase 2 trials, shaping the next wave of combination therapies aimed at defending against cardiometabolic decline 60.
| Therapeutic Candidate | Mechanism of Action | Target Indication / Pathology | Clinical Stage (as of 2026) |
|---|---|---|---|
| Dasatinib + Quercetin | Senolytic (Tyrosine Kinase + Flavonoid) | Clearance of senescent cells, SASP reduction | Phase 2 (Diabetic Kidney Disease) |
| UBX1325 | Senolytic (BCL-xL Inhibitor) | Localized clearance of senescent vascular cells | Phase 2 (Diabetic Macular Edema) |
| BGE-102 | Oral NLRP3 Inflammasome Inhibitor | Systemic inflammation, CV risk, ocular disease | Phase 2 (CV Risk / DME initiating mid-2026) |
| Rapamycin | mTOR Inhibitor (Nutrient Sensing) | Immunosenescence, autophagic decline | Phase 2 (Healthy Aging / Immunomodulation) |
| Tirzepatide | GLP-1/GIP Receptor Agonist | Obesity, Type 2 Diabetes, metabolic stress | FDA Approved (Ongoing Combo Trials) |
| Azelaprag (BGE-105) | APJ Receptor Agonist | Obesity, muscle atrophy | Phase 2 (STRIDES trial halted 2024) |
| BGE-117 | HIF-PH Inhibitor | Hypoxia signaling, Unexplained Anemia | Phase 2a (Unexplained Anemia of Aging) |
Global Initiatives and Future Clinical Translation
The successful clinical translation of vascular aging therapeutics requires a paradigm shift in how aging is measured, funded, and evaluated by regulatory bodies. Because chronological life extension trials require decades to complete, the field relies heavily on the validation of the surrogate biomarkers (such as epigenetic clocks and hsCRP levels) discussed previously, capable of capturing the rate of biological aging in a contracted timeframe 3447.
The XPRIZE Healthspan and Philanthropic Investment
The momentum toward clinical translation is heavily supported by massive philanthropic and global institutional initiatives. The Hevolution Foundation, a prominent backer of aging biology based in Riyadh, has allocated over $400 million to healthspan science, bridging the critical funding "valley of death" that often prevents Phase II clinical translation 6162. Investment in the broader longevity domain surged to over $7.3 billion in early 2024, reflecting a maturation of the field toward later-stage clinical trials 61.
In parallel, the $101 million XPRIZE Healthspan competition, launched in 2023, represents a highly structured global effort to identify therapeutics that can safely and accessible restore muscle, cognitive, and immune function by a minimum of 10 years in adults aged 50-80 6364. As of mid-2025, semifinalist teams have been actively advancing into early-stage clinical trials, with the rigorous requirement that winning interventions must demonstrate real-world, multi-systemic efficacy within a single year of treatment 626465. Competitors encompass a wide range of modalities; for instance, Longeveron was named a Top 40 Semifinalist based on the clinical feasibility of laromestrocel (Lomecel-B), an allogeneic mesenchymal stem cell therapy evaluated for its pro-vascular, anti-inflammatory, and regenerative effects in Alzheimer's disease and aging-related frailty 77. The finalists' human clinical trials, overseen by the University of Utah Data Coordinating Center, will run from 2026 to 2029 6465.
Evolving Cardiovascular Treatment Guidelines
Concurrently, major medical societies are formally integrating the biology of aging into clinical cardiovascular practice. The 2025 and 2026 American Heart Association (AHA) and European Society of Cardiology (ESC) consensus statements explicitly emphasize that age-related cardiovascular changes - such as arterial stiffness and endothelial dysfunction - require tailored, biology-centered care rather than reliance on chronological age alone 406667.
These evolving guidelines advocate for earlier, intensive risk factor management. For primary prevention, the AHA heavily emphasizes utilizing subclinical disease measures, such as Coronary Artery Calcium (CAC) scoring. In adults with a CAC score of $\ge$1000 AU, intensive lipid-lowering therapies (LLT) are mandated to achieve a target LDL-C of <55 mg/dL, while a score of $\ge$300 to 999 AU warrants a target of <70 mg/dL 68. To achieve these deep reductions, the integration of novel therapeutics beyond statins, such as PCSK9 inhibitors (e.g., evolocumab), is recommended to drastically lower lipid burdens and mitigate the progression of atherosclerotic vascular aging 6769.
Furthermore, the guidelines emphasize shared decision-making frameworks that account for geriatric syndromes, cognitive function, and individual healthspan goals when considering complex interventions like revascularization in older adults 40. By shifting the clinical focus from the reactive treatment of end-stage ischemic events to the proactive preservation of vascular resilience, the integration of advanced gerotherapeutics, precision biomarkers, and updated clinical protocols holds the potential to fundamentally decouple chronological aging from cardiovascular decline.