Mechanisms of physiological brain aging
Conceptual Framework of Cognitive Aging
The human central nervous system undergoes continuous structural, functional, and molecular transformations across the lifespan. Historically, research conflated age-related cognitive decline with early-stage pathological neurodegeneration. However, physiological brain aging is a distinct biological trajectory characterized by a gradual decline in select cognitive domains - such as cognitive processing speed, working memory, and executive function - while maintaining largely intact activities of daily living and semantic knowledge 12. Pathological neurodegeneration, encompassing conditions such as Alzheimer's disease (AD) and Parkinson's disease (PD), involves catastrophic cellular loss, extensive brain atrophy, and the accumulation of neurotoxic protein aggregates 34.
In stark contrast, normal cognitive aging is driven by subtle, widespread, and interconnected alterations in synaptic integrity, epigenetic regulation, glial state transitions, white matter microstructure, and cerebrovascular hemodynamics 34. Isolating these mechanisms requires examining the specific variables that mediate biological decline independently of overt disease. This analysis explores the multi-scalar determinants of physiological brain aging, moving from intracellular molecular networks to systemic physiological influences.
Cellular Proteostasis and Mitochondrial Dynamics
Proteostatic Collapse
At the intracellular level, physiological aging is precipitated by a progressive decline in protein homeostasis (proteostasis). Proteostasis relies on a tightly regulated equilibrium between protein synthesis, folding, and degradation. Experimental models of accelerated aging, such as the turquoise killifish (Nothobranchius furzeri), demonstrate that normal aging intrinsically disrupts the central machinery responsible for protein manufacturing 5. During physiological aging, there are observable dysregulations in amino acid concentrations, transfer RNA (tRNA) availability, and messenger RNA (mRNA) translation dynamics, leading to increased aggregation and dysfunction in synthesis processes 5.
Unlike AD, where specific hallmark proteins (amyloid-beta and hyperphosphorylated tau) form dense extracellular plaques and intracellular tangles, normal aging involves a generalized, low-grade accumulation of misfolded proteins. This occurs due to age-related impairments in the unfolded protein response (UPR) and the induction of chronic endoplasmic reticulum (ER) stress 67. Furthermore, nutrient-sensing pathways that govern cellular metabolism undergo significant shifts; while the mechanistic target of rapamycin (mTOR) signaling pathway and growth hormone levels naturally decrease during physiological aging, pathological aging is often associated with hyperactivated mTOR pathways and localized brain insulin resistance 3.
Mitochondrial Dysfunction and Oxidative Stress
Mitochondrial integrity is central to maintaining the high bioenergetic demands of neural tissue. Because the brain accounts for a disproportionate amount of reactive oxygen species (ROS) consumption, it exhibits extreme susceptibility to oxidative stress 8. In normal aging, neural mitochondria undergo profound structural modifications, including morphological enlargement and progressive fragmentation, alongside a measurable reduction in oxidative phosphorylation and the accumulation of mitochondrial DNA (mtDNA) mutations 3.
The age-related impairment of mitophagy - the selective macroautophagic clearance of damaged mitochondria - results in severe bioenergetic insufficiency 9. The consequent overproduction of ROS damages critical cellular components, including genomic DNA, leading to genomic instability. In normal aging, accumulated DNA damage predominantly affects the promoter regions of genes associated with synaptic plasticity, mitochondrial function, and neuronal survival, thereby diminishing their baseline expression 3.
Genomic Instability and Epigenetic Clocks
Epigenetic Modifications in Neural Tissue
Epigenetic modifications provide a mechanism by which environmental, metabolic, and chronological factors influence brain aging without altering the underlying DNA sequence. Normal brain aging is associated with highly specific epigenetic shifts, including an increase in histone methylation and a concurrent decrease in histone acetylation in the promoter regions of synaptic genes 3. For example, healthy aging is associated with a significant accumulation of the histone marker H4K16ac, whereas pathological neurodegeneration entails a marked loss of H4K16ac near genes essential for cellular survival 3.
DNA Methylation Clocks and Biological Age Acceleration
Advancements in DNA methylation (DNAm) have enabled the development of "epigenetic clocks" that quantify biological aging as distinct from chronological aging. First-generation clocks (e.g., Horvath and Hannum) were trained strictly on chronological age, while second- and third-generation clocks, such as DNAm PhenoAge, GrimAge, AgeAccelGrim2, and DunedinPACE, incorporate physiological biomarkers and mortality risk factors to estimate the precise pace of biological decline 1011.
Accelerated biological aging, as measured by these advanced clocks, strongly correlates with structural brain atrophy, reduced cognitive fitness, and a statistically higher risk of developing mild cognitive impairment (MCI) 1112. Longitudinal cohorts, such as the Study of Latinos-Investigation of Neurocognitive Aging (SOL-INCA) comprising 2,671 participants, demonstrate that between-visit increases in age acceleration for GrimAge and PhenoAge are significantly associated with a greater risk of global cognitive decline 1314.
The application of machine learning to 1,397 healthy human cortex samples has further refined these tools, yielding brain-specific epigenetic clocks based on 347 distinct methylation sites calibrated specifically to central nervous system tissue 15. Utilizing these cell-type resolution clocks, researchers have discovered that different neural cell populations age at disparate rates. Glial cells, for instance, exhibit accelerated epigenetic aging compared to neurons, suggesting that non-neuronal cells may act as primary drivers of the epigenetic aging trajectory within the brain 16.
Synaptic Degradation and Structural Connectivity
Debunking the Neuronal Cell Death Model
A pervasive and historical misconception regarding brain aging is the assumption that the process inevitably involves massive, widespread neuronal cell death. Modern neuroanatomical studies and stereological counting methods have conclusively debunked this notion. If an individual ages without developing a neurodegenerative disorder, they do not lose a statistically significant number of cortical neurons 417. Postmitotic, terminally differentiated neurons generally survive throughout the human lifespan under normal physiological conditions 67.
Instead, the cognitive decline observed in healthy aging is predominantly driven by alterations at the synapse 47. Normal aging is characterized by a significant reduction in dendritic branching, decreased spine density, and the functional downregulation of synaptic genes, particularly within the prefrontal cortex and the hippocampus 3.
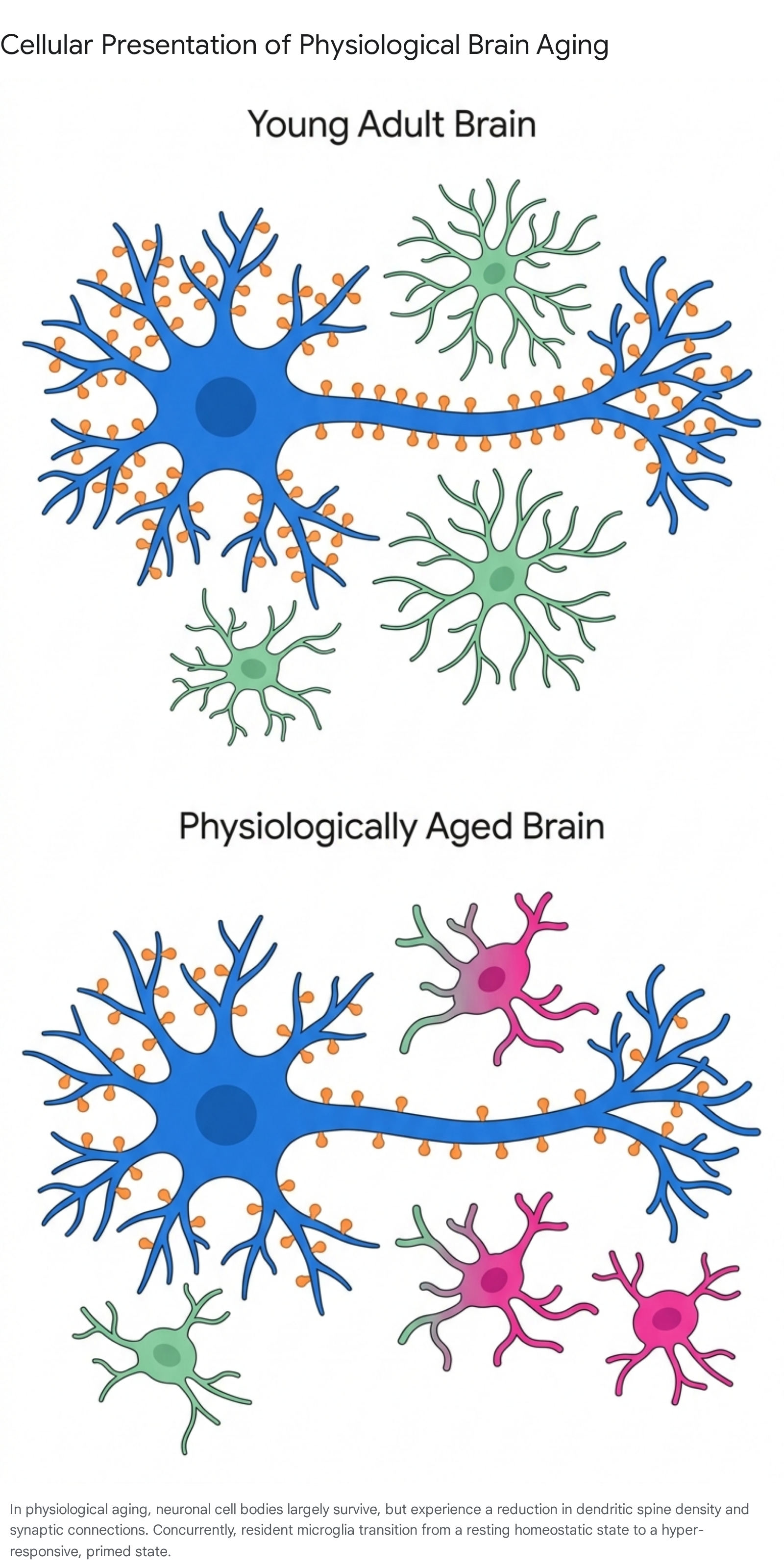
The disruption of intercellular communication and micro-circuit integrity, rather than the outright apoptosis of neuronal cell bodies, accounts for observed deficits in working memory, attention, and executive function 3. This distinction is therapeutically critical: because the neuronal soma remains alive in physiological aging, interventions aimed at restoring synaptic health and plasticity possess a viable target, whereas reversing cell death in advanced neurodegeneration remains an insurmountable barrier 417.
The Adult Hippocampal Neurogenesis Debate
The functional relevance of adult hippocampal neurogenesis (AHN) in humans remains one of the most fiercely debated topics in neurobiology. While it is well-established that neurogenesis persists in the dentate gyrus of adult rodents, providing a continuous source of new neurons that integrate into memory circuits, evidence in humans is highly contested 617.
The advent of single-cell and single-nucleus RNA sequencing (sc/snRNA-seq) was initially anticipated to resolve this controversy by identifying precise transcriptomic signatures of neurogenic populations. However, recent studies have yielded profoundly conflicting conclusions 1819. Certain profiling studies have identified sparse populations of cells with neural stem cell (NSC) or immature neuronal characteristics in the adult human hippocampus 1820. Conversely, rigorous cross-species comparisons have reported a sharp developmental drop in neurogenesis during infancy, reaching undetectable levels in adult human tissue 17.
These contradictions stem from several profound methodological and conceptual challenges. First, traditional immunohistochemical markers for immature neurons, such as doublecortin (DCX), are not exclusively specific to neurogenic lineages in humans. DCX expression has been detected in non-neurogenic cell types, including mature astrocytes and interneurons, leading to false positives 20. Second, adult radial glial-like cells (RGLs), the purported NSCs, exhibit transcriptomic profiles that overlap heavily with mature astrocytes. Identifying human-specific marker genes to segregate these populations remains difficult, as there is minimal overlap between human and rodent neurogenic markers 18.
Finally, the quality of postmortem human brain tissue introduces significant confounding variables. Postmortem delay (PMD), tissue processing methodologies, and the inherent neuroinflammatory state of the donor prior to death can significantly alter RNA stability and marker expression 1718. Given these constraints, current consensus reflects high uncertainty, suggesting a highly restricted or negligible rate of de novo AHN in adult humans. This indicates that the human hippocampus relies primarily on other forms of synaptic plasticity, rather than the generation of new neurons, to maintain cognitive function during aging 1720.
Microglial Trajectories and State Transitions
From Homeostasis to Dystrophy
Microglia, the resident innate immune macrophages of the central nervous system, act as the primary surveyors of the brain microenvironment. During youth, microglia exist in a homeostatic state characterized by a highly ramified morphology, executing vital functions such as synaptic pruning, tissue repair, and debris phagocytosis 2122. However, physiological aging induces a progressive, irreversible loss of this homeostatic profile.
As the brain ages, microglia transition into intermediate, stress-responsive states before adopting a fully "primed" or dystrophic phenotype 2123. Single-cell RNA-sequencing and pseudotime trajectory analyses have intricately mapped these intermediate states. Initially, mitochondrial dysfunction and systemic inputs trigger a stress response, leading to the transient upregulation of immediate early genes (e.g., Jun, Fos, Klf2) and the purinergic receptor gene P2ry12 2425. Products of this stress response, notably transforming growth factor beta 1 (TGFβ1), operate through autocrine signaling mechanisms in an attempt to return the microglial cells to homeostasis 212326.
The Phenotype of the Primed Microglia
Over time, this compensatory stress response fails. Aged microglia experience a downregulation of essential homeostatic transcription factors (such as RUNX1, IRF8, and PU.1) and an upregulation of genes associated with innate immune activation, including B2m, Apoe, Cd48, Lyz2, and major histocompatibility complex (MHC) presentation pathways 2427.
Morphologically, these cells become dystrophic - exhibiting deramified, thickened, and retracted processes 22. Functionally, primed microglia exhibit severely diminished phagocytic capacity but become hyper-reactive to secondary inflammatory stimuli. They chronically release pro-inflammatory cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α) 2227.
This microglial activation is not uniform across the brain. Quantitative analyses of the lysosomal marker CD68 and the pro-inflammatory transcription factor NFKB p65 reveal striking spatial heterogeneity within the aging hippocampus. Regions such as the granule cell layer (GC), hilus, CA3, and outer CA1 show robust, age-related microglial activation, whereas the molecular layer (ML) and inner CA1 remain relatively devoid of these age-related inflammatory changes 23. This chronic, low-grade, region-specific neuroinflammation - often termed "inflammaging" - directly contributes to the synaptic dysfunction underlying cognitive decline, independent of specific disease pathology 212225.
Cerebrovascular Integrity and Fluid Dynamics
Blood-Brain Barrier Permeability
The neurovascular unit (NVU) - comprising cerebral endothelial cells, pericytes, astrocytes, and the basement membrane - forms the blood-brain barrier (BBB), a highly selective physiological filter maintaining brain homeostasis 2829. Normal aging exerts a profound, deleterious impact on both the structural and functional integrity of the BBB.
Age-related BBB breakdown is initiated by the reduced expression of key tight junction proteins, specifically claudin-5, occludin, and zonula occludens-1 (ZO-1) 828. Concurrently, the capillary network undergoes morphological degradation, involving the detachment of pericytes, the swelling of astrocytic end-feet, and a reorganization of the endothelial actin cytoskeleton. This cellular disruption is heavily driven by chronic oxidative stress acting through the RhoA, PI3, and PKB/Akt signaling pathways 8.
Beyond paracellular tight junction failure, physiological aging disrupts tightly regulated transcellular transport mechanisms. There is a documented age-dependent downregulation of major facilitator superfamily domain-containing protein 2A (MFSD2A), a critical lipid transporter that normally suppresses transcytosis 2829. The loss of MFSD2A derepresses caveolin-1, leading to an abnormal increase in non-specific vesicular transport across the endothelium. Additionally, aging reduces the expression of essential transporters like GLUT1 (affecting glucose delivery), LRP1, and P-glycoprotein, while upregulating RAGE receptors 28.
Consequently, neurotoxic blood-derived proteins (such as albumin and fibrinogen) leak into the brain parenchyma, provoking localized neuroinflammation and vasogenic edema. These microvascular leakages are detectable as white matter hyperintensities on dynamic contrast-enhanced magnetic resonance imaging (MRI). Because BBB permeability frequently manifests earlier than observable structural brain atrophy, microvascular dysfunction serves as a primary, upstream driver of emerging cognitive impairment 830.
The Glymphatic System and Fluid Clearance
The glymphatic system is a brain-wide perivascular network responsible for the exchange of subarachnoid cerebrospinal fluid (CSF) and brain interstitial fluid (ISF), facilitating the clearance of metabolic waste products, including soluble amyloid-beta, tau, and lactate 3132. This convective bulk flow is primarily mediated by aquaporin-4 (AQP4) water channels densely localized on the astrocytic end-feet that ensheath the cerebrovasculature 3233.
Recent advanced neuroimaging techniques, specifically diffusion-based intravoxel incoherent motion (IVIM) and perfusion-based multi-echo arterial spin labeling (ASL), have successfully differentiated the specific effects of physiological aging from those of sleep disruption on human glymphatic dynamics 31.
Research indicates that older adults exhibit significantly reduced cortical cerebral blood flow (CBF) and a shortened trans-vascular water exchange time (Tex) between the vasculature and the perivascular space, pointing to an impaired fluid transfer efficiency into the brain parenchyma 31. Interestingly, while acute sleep deprivation primarily reduces the mobility of CSF within the large subarachnoid spaces (measured by pseudodiffusion coefficient, D*), the microvascular perfusion deficits and altered transendothelial exchange are uniquely driven by chronological aging 31. The impairment of this fluid transport system over decades results in a sluggish clearance of metabolic byproducts, contributing heavily to the sterile neuroinflammation and proteostatic collapse characteristic of the aging brain 3234.
Intracortical Myelin Degradation
Myelin, the lipid-rich sheath insulating axonal tracts, is critical for the rapid transduction of action potentials and the precise synchrony of neural networks. The myelination of the human cerebral cortex is not a static developmental event limited to childhood, but a lifelong plastic process responsive to learning and memory 35. Utilizing ultra-high field (7 Tesla) quantitative MRI to measure longitudinal relaxation rates (R1) and transverse relaxation rates (R2), researchers have accurately mapped the trajectory of intracortical myelin across the human lifespan 3536.
The trajectory of cortical myelination follows a distinct quadratic, inverted-U shape. Myelin volume fractions increase progressively through adolescence and early adulthood, reaching a global peak in the late 30s to early 40s, before undergoing a protracted decline beginning in the fifth and sixth decades of life 363738.
Crucially, this age-related demyelination is anatomically heterogeneous. The precise timeline of myelin maturation and eventual degradation heavily depends on the functional role of the specific cortical region, as outlined in the data comparison below.
Comparative Timelines of Regional Myelin Density
| Cortical Region | Maturation Profile | Peak Age of Myelin Density | Age-Related Degradation Pattern |
|---|---|---|---|
| Visual Cortex (e.g., Splenium) | Early-myelinating | ~34 years | Remains relatively stable during normal aging; highly resistant to breakdown 363940. |
| Motor & Premotor Areas | Intermediate-myelinating | ~38 years | Gradual decline following peak; follows standard global brain averages 3640. |
| Prefrontal Cortex (e.g., Genu) | Late-myelinating | Early to Mid 40s | Exceptionally vulnerable; undergoes the steepest and most significant age-related degradation 3639. |
Because the prefrontal cortex governs higher-order executive function, working memory, and cognitive processing speed (CPS), the degradation of myelin integrity in this specific topography acts as a direct biological mediator of age-related cognitive slowing. Statistical models confirm that the breakdown of late-myelinating white matter (LMWM) mediates the relationship between advanced age and slowing CPS, indicating that myelin loss is a primary structural mechanism driving normative cognitive decline 3539.
Systemic Milieu and Heterochronic Parabiosis
The Influence of Circulating Factors
The brain does not age in isolation; it is continuously exposed to a systemic milieu of circulating proteins, lipids, and extracellular vesicles. The profound influence of these systemic factors on brain aging has been elucidated through heterochronic parabiosis - a surgical experimental model wherein the circulatory systems of young and old animals are conjoined to share a common blood supply 414243.
These studies conclusively demonstrate that the systemic environment is capable of driving or reversing cellular aging mechanisms. Exposing an aged brain to young blood can restore BBB integrity, enhance neurovascular coupling, increase capillary density, and reduce the burden of senescent cells 414344. Conversely, exposing a young animal to aged blood rapidly induces vascular dysfunction, reduces neurogenesis, and triggers neuroinflammation, driven heavily by pro-geronic factors circulating in older plasma, such as CCL11, osteopontin, and beta-2 microglobulin (B2M) 4142.
The GDF11 Controversy and Novel Rejuvenating Factors
The search for specific "rejuvenating" anti-geronic factors has yielded several candidates, though the field is marked by ongoing controversy. Growth Differentiation Factor 11 (GDF11), a member of the TGF-β superfamily, was initially identified as a potent systemic factor that declines with age. Early reports suggested that exogenous administration of recombinant GDF11 could reverse age-related cardiac hypertrophy and stimulate neurogenesis and blood vessel density in the aging brain 454648.
However, subsequent studies generated highly conflicting results regarding its safety, efficacy, and its true age-related trajectory. A major source of this controversy lies in the biochemical structure of GDF11, which shares 90% homology with GDF8 (myostatin), differing by only 11 amino acids in their mature signaling domains 4547. Because many commercial antibodies cross-react with both proteins, initial measurements of age-related decline were called into question. While recent rigorous assays confirm that GDF11 is biochemically distinct and significantly more potent at inducing SMAD2 phosphorylation than GDF8, the dose-dependent effects and exact therapeutic viability of GDF11 for human brain rejuvenation remain unresolved 4547.
More recently, pigment epithelium-derived factor (PEDF) has emerged as a novel, less contested circulating protein that declines with mammalian aging. Utilizing in vitro heterochronic parabiosis screening systems, researchers discovered that PEDF extended the replicative lifespan of primary human fibroblasts. When systemically administered to aged mice, PEDF reversed age-related functional decline across multiple tissues and measurably improved cognitive function, positioning it as a highly promising mediator of systemic homeostasis 44.
Diagnostic Biomarkers of Neurodegeneration Versus Aging
Differentiating physiological brain aging from the preclinical stages of neurodegeneration is a critical clinical challenge. While normal aging features generalized structural atrophy and mild memory changes, pathological aging is defined by the localized, toxic accumulation of specific misfolded proteins.
Recent breakthroughs in blood-based and CSF biomarkers allow for highly sensitive, minimally invasive stratification of these divergent trajectories. The table below outlines the differential behavior of primary biomarkers in normal aging versus early Alzheimer's disease.
| Biomarker Profile | Physiological Brain Aging | Preclinical / Early Neurodegeneration (AD) |
|---|---|---|
| Plasma p-tau217 & p-tau231 | Stable; remains well below established diagnostic thresholds (e.g., < 0.09 pg/mL) 48. | Highly elevated; indicates active tau phosphorylation and correlates strongly with early amyloidosis before PET positivity 4849. |
| Neurofilament Light (NfL) | Gradual, subtle increase reflecting normal, age-related myelin and axonal turnover 5051. | Sharp, significant elevation reflecting acute neuroaxonal injury and rapid neuronal death 5051. |
| Aβ42 / Aβ40 Ratio (CSF) | Remains normal or shows minor, non-pathological shifts reflecting steady clearance 5253. | Significantly decreased, indicating abnormal brain retention and the formation of Aβ plaques 5253. |
| Glial Fibrillary Acidic Protein (GFAP) | Mild elevation corresponding to normal microglial priming and stress responses 51. | Highly elevated, indicating severe, chronic astrogliosis and advanced neuroinflammation 51. |
Plasma phosphorylated tau 217 (p-tau217) and p-tau231 have emerged as the most specific indicators of early Alzheimer's disease pathology, rising significantly even in the preclinical, cognitively unimpaired stages 484950. P-tau217 strongly correlates with early neocortical amyloid-beta (Aβ) accumulation and can accurately distinguish AD from typical age-related cognitive decline with high sensitivity and specificity 4849. In contrast, Neurofilament light chain (NfL) is a highly sensitive but disease-agnostic marker of neuroaxonal injury; it will rise in AD, ALS, or following a stroke, making it an excellent measure of generalized brain damage but insufficient for diagnosing specific proteinopathies alone 495051.
Global Longitudinal Cohorts and Cognitive Resilience
Despite facing highly similar biological and molecular changes, individual humans exhibit vastly different cognitive trajectories as they age. The study of these diverse outcomes relies heavily on massive, multi-decade global longitudinal cohorts. Prominent examples include the Chinese Longitudinal Healthy Longevity Survey (CLHLS), the Health and Aging in Africa: A Longitudinal Study of an INDEPTH Community in South Africa (HAALSA), the Study of Latinos-Investigation of Neurocognitive Aging (SOL-INCA), and the Ibadan Study of Aging in Nigeria 1354555657.
Group-based trajectory modeling applied to these diverse populations consistently demonstrates that older adults segregate into distinct psychological resilience groups: persistent high resilience, persistent middle resilience, and decreasing resilience 5455. Individuals whose psychological resilience declines over time experience a drastically higher hazard ratio (HR = 2.46 in the CLHLS cohort) for developing cognitive impairment, entirely independent of their baseline chronological age or basic biological markers 5558.
Factors such as higher educational attainment, the rigorous clinical management of vascular risk factors (e.g., hypertension and obstructive sleep apnea), and consistently low depression indices interact directly with the brain's biological state 305960. These lifestyle and psychological factors build "cognitive reserve," mitigating the neurotoxic effects of microvascular damage, counteracting the slowing of cognitive processing speed caused by prefrontal myelin degradation, and preserving high-level executive function throughout the physiological aging process 3059.