Information theory of aging and epigenetic noise
The precise molecular mechanisms driving the physiological and cellular decline associated with advanced age remain a subject of intense scientific inquiry. For decades, the prevailing paradigm positioned random somatic mutations and irreversible DNA damage as the primary, upstream drivers of cellular aging. Under this traditional framework, aging is viewed as a hardware problem - an inevitable, unidirectional accumulation of structural defects in the genetic code. However, a growing body of evidence suggests that aging may be fundamentally driven by a loss of epigenetic regulation rather than strict genetic sequence degradation.
The Information Theory of Aging (ITOA), formalized in recent years by researchers including David Sinclair, provides a comprehensive framework proposing that biological aging is the result of a progressive loss of youthful epigenetic information 12. This theory suggests that the "software" of the cell - the epigenome - becomes corrupted over time due to cellular stress, leading to a loss of cellular identity and widespread tissue dysfunction. Crucially, the ITOA posits that cells retain a latent "backup copy" of their youthful epigenetic state, which can be theoretically and experimentally accessed to reverse the aging process 22. This paradigm shift moves aging from an inevitable accumulation of physical damage to a potentially reversible loss of information.
Theoretical Foundations of Biological Information
To understand the Information Theory of Aging, it is necessary to precisely distinguish between the two primary modalities of biological information storage within the eukaryotic cell: the genome and the epigenome.
The genome acts as a foundational blueprint composed of nucleic acids. It is characterized by its highly stable, digital nature, operating sequentially through discrete nucleotide pairings (adenine, cytosine, thymine, and guanine) 12. Genetic information is replicated during cell division with remarkably high fidelity, relying on robust error-checking mechanisms by DNA polymerases to maintain the integrity of the code.
The epigenome, conversely, consists of the physical and chemical modifications superimposed upon the DNA and its packaging structures. These include chemical modifications directly to the DNA (such as cytosine methylation at CpG dinucleotides) and post-translational modifications to the histone proteins around which DNA is wrapped (such as methylation, acetylation, phosphorylation, and ubiquitination) 23. Furthermore, the epigenome encompasses the three-dimensional architecture of chromatin within the nucleus, dictated by chromatin-remodeling complexes and non-coding RNAs 245. These epigenetic marks dictate precise gene expression patterns, ensuring that a post-mitotic neuron behaves strictly as a neuron and a hepatocyte as a hepatocyte, despite both containing the exact identical genetic code 12.
Unlike the highly stable genetic code, epigenetic information is stored in a format described as "digital-analog" 12. This continuous, responsive format makes the epigenome highly sensitive to environmental inputs, metabolic fluctuations, and cellular stress, allowing organisms to adapt rapidly to changing conditions. However, this same plasticity renders the epigenome inherently susceptible to degradation, drift, and accumulated noise over long biological timescales 12.
Shannon Entropy and Biological Mapping
The conceptual framework of the ITOA draws directly from the 1948 mathematical theory of communication developed by communications engineer and mathematician Claude Shannon 26. In Shannon's foundational model, a signal is transmitted from an information source to a receiver through a channel. However, transmission is inevitably obscured by a noise source, requiring a correcting device or an observer to verify and preserve signal integrity. The ITOA maps these abstract mathematical components directly to mammalian biology to explain the entropy of aging.
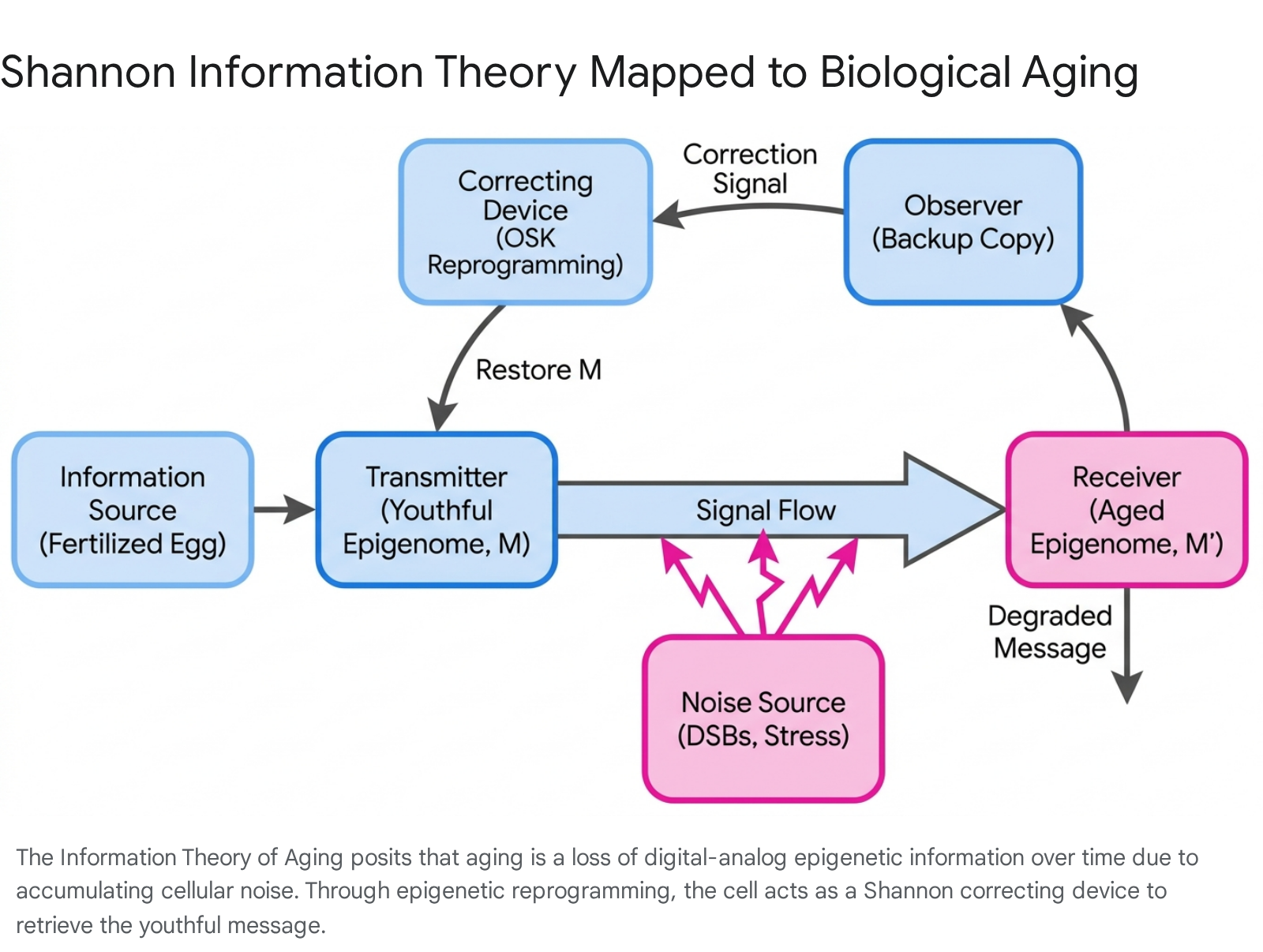
Within the ITOA framework: * The Youthful Message (M): This represents the pristine, highly ordered epigenetic landscape established during embryonic development. This initial state defines optimal cellular identity, robust function, and youthful gene expression 26. * The Aged Message (M'): The degraded, corrupted epigenetic signal that results from decades of molecular interference. This manifests as altered gene expression and lost cellular identity 26. * The Noise Source: The primary drivers of epigenetic entropy. This includes cellular damage, continuous DNA double-strand breaks (DSBs), physical trauma, viral infections, oxidative stress, and general environmental toxicity 267. * The Observer (Backup Copy): An embedded repository of youthful epigenetic information retained within the somatic cell. This observer "notes" the original intended state and survives the aging process, theoretically capable of recognizing the deviation from the pristine signal 2. * The Correcting Device: The molecular machinery involved in epigenetic reprogramming (such as the forced expression of embryonic transcription factors). When activated, this device accesses the observer's backup copy to restore the youthful message, overcoming the accumulated noise 26.
Waddington's Landscape and Epigenetic Drift
The degradation of the youthful message is frequently conceptualized using the metaphor of Waddington's epigenetic landscape. In developmental biology, cellular differentiation is visualized as a marble rolling down a contoured hill 89. At the top is the pluripotent stem cell; as the marble rolls downward, it enters specific valleys representing specialized cellular fates (e.g., a muscle cell or a lymphocyte). The steep ridges between these valleys are constructed by the epigenome, locking the cell securely into its differentiated state 89.
According to the ITOA, the accumulation of epigenetic noise gradually erodes these ridges. As the structural integrity of the epigenome weakens over decades, the valleys become shallow. The marbles (cells) begin to drift laterally, losing their tight phenotypic constraints. This epigenetic drift results in an older cell exhibiting a confused identity - a phenomenon that forms the core pathology of aging at the single-cell level 8910.
Molecular Mechanisms of Epigenetic Noise
The central mechanistic driver of the ITOA is the Relocalized Chromatin Modifier (RCM) hypothesis, which dictates how exactly this epigenetic noise accumulates. The RCM hypothesis suggests that the epigenetic landscape erodes primarily as an inadvertent consequence of the cell's own emergency DNA repair mechanisms 214.
The genome is continuously subjected to damage from endogenous metabolic byproducts, such as reactive oxygen species, and exogenous hazards like ultraviolet radiation and chemical toxins 11. The most severe form of this damage is the DNA double-strand break (DSB) 311. To survive a DSB, the cell must initiate an immediate, highly coordinated emergency response to repair the genome and prevent apoptosis or malignant transformation.
The Emergency Response Pathway
When a DSB occurs, the highly compacted chromatin surrounding the break must be temporarily unwound to allow the physical access of repair enzymes (such as those involved in homologous recombination or non-homologous end joining) 711. This process requires the rapid mobilization of numerous epigenetic modifiers, which must temporarily abandon their usual genomic loci - where they normally maintain stable gene expression - to assist at the site of the break 2712.
The initiation of this sequence begins with the phosphorylation of the histone variant H2AX ($\gamma$H2AX) at the damage site, serving as a beacon for repair machinery 3713. Simultaneously, specific histone acetyltransferases and deacetylases modify the local chromatin. A critical player in this response is the NAD+-dependent deacetylase SIRT1, a mammalian ortholog of the yeast Sir2 protein long associated with longevity 2713.
SIRT1, alongside members of the Polycomb Repressive Complex 2 (PRC2) such as EZH2, and DNA methyltransferases like DNMT1 and DNMT3B, rapidly migrate to the break 712. At the site, SIRT1 removes acetyl groups from histone H4 at lysine 16 (H4K16ac), while EZH2 facilitates the deposition of the repressive mark H3K27me3 7. These modifications rapidly re-compact the chromatin surrounding the break, suppressing local transcription to prevent the production of truncated or mutated RNA transcripts from the damaged template 7.
| Pathway Stage | Molecular Action | Epigenetic Consequence |
|---|---|---|
| 1. Damage Recognition | Double-strand break (DSB) occurs. | Phosphorylation of H2AX ($\gamma$H2AX) signals the damage site 713. |
| 2. Chromatin Relaxation | Transient acetylation of H4K16. | Localized chromatin unwinds to allow repair protein access 7. |
| 3. Factor Relocalization | SIRT1, EZH2, and DNMTs abandon native loci. | Original target genes temporarily lose their regulatory silencing controls 712. |
| 4. Repair Compaction | SIRT1 deacetylates H4K16; EZH2 enriches H3K27me3. | Chromatin around the break is tightly compacted to halt local transcription during repair 7. |
| 5. Epigenetic Drift | Imperfect return of factors to native loci. | In a fraction of events, factors fail to perfectly reset, leading to persistent noise and altered methylation 7. |
Once the DNA sequence is successfully ligated, these chromatin-modifying proteins must return precisely to their original positions to restore the cell's baseline epigenetic state. Experimental evidence indicates that in the vast majority of cases (approximately 99.1%), this restoration is successful, and the chromatin returns to its original configuration 7. However, in a minute fraction of events (roughly 0.9%), the silencing factors fail to return correctly, or they permanently associate with the break site, establishing a persistent "epigenetic memory of silencing" where there was none before 7.
Over the course of a mammalian lifespan, the sheer volume of billions of repair events leads to an accumulation of imperfect molecular repositioning. This slow, steady accumulation of incorrectly localized epigenetic marks constitutes the "epigenetic noise" that obscures the youthful message 218.
Heterochromatin Loss and Inflammaging
The macroscopic consequence of this accumulating noise is a structural deterioration of the genome, characterized heavily by heterochromatin loss. Chromatin exists broadly in two states: euchromatin, which is loosely packed and transcriptionally active, and heterochromatin, which is tightly condensed, highly methylated, and transcriptionally silenced 2414.
Youthful chromatin is characterized by distinctly partitioned regions of tightly packaged heterochromatin, maintained by repressive histone marks (such as H3K9me3 and H3K27me3) and heterochromatin protein 1 (HP1) binding 41516. During aging, the persistent reallocation of factors like SIRT1 and SIRT6 away from their native loci results in a global unraveling of these heterochromatic domains 417. This is accompanied by a global hypomethylation of the genome alongside focal areas of aberrant hypermethylation, particularly at tumor suppressor gene promoters 41417.
The unspooling of heterochromatin has profound downstream effects. Most notably, it leads to the derepression of transposable elements (TEs) such as Long Interspersed Nuclear Elements (LINE-1) 917. Normally kept strictly silenced in healthy somatic tissues, these ancient viral remnants become transcriptionally active as heterochromatin barriers fall 917. When activated, these retrotransposons can self-amplify and insert themselves randomly into the genome, driving insertional mutagenesis and severe genomic instability 917.
Furthermore, the presence of cytosolic DNA from active transposons triggers innate immune pattern recognition receptors. This drives a chronic, low-grade, sterile inflammatory state commonly referred to as "inflammaging," which accelerates systemic tissue decline and is implicated in neurodegeneration and cellular senescence 1718.
Cellular Ex-Differentiation
As the strict boundaries between euchromatin and heterochromatin erode, the precise gene regulatory networks of the cell begin to fail. Genes that should be permanently silenced in a specific adult tissue type are inadvertently transcribed 161920. This phenomenon, termed "ex-differentiation" or "dys-differentiation," represents the functional endpoint of epigenetic noise 21921.
Because the cell no longer has the correct epigenetic tags to suppress inappropriate genetic programs, it loses its specialized identity. A skin cell may begin exhibiting characteristics of other lineages; a mature neuron may lose its strict synaptic clarity; stem cells lose their regenerative potential and fall into senescence 21819. The ITOA argues that this systemic cellular confusion, multiplied across billions of cells within an organ, is the primary etiology of age-related diseases.
Epigenetic Clocks and Methylation Entropy
The predictable nature by which the epigenome degrades has allowed researchers to construct powerful biomarkers of aging known as epigenetic clocks. Developed extensively over the last decade by researchers such as Steve Horvath, these predictive algorithms estimate a subject's biological age by analyzing specific DNA methylation patterns 222823.
Epigenetic clocks measure the accumulation of methyl groups at specific cytosine-phosphate-guanine (CpG) dinucleotide regions 2228. Over time, the genome exhibits a predictable, systematic drift: some regions become heavily hypermethylated, while others undergo widespread hypomethylation 428. First-generation clocks were trained purely on chronological age, while second-generation clocks (such as PhenoAge and GrimAge) incorporate clinical biomarkers, lifestyle factors, and mortality risk to provide a more accurate reflection of physiological healthspan and the pace of biological aging 1722.
It is important to draw a scholarly distinction between "epigenetic noise," "heterochromatin loss," and "epigenetic clocks." While deeply related, they represent different facets of the aging paradigm: * Heterochromatin Loss refers to the physical unspooling of tightly packed genomic regions, leading to transposon activation and gross structural changes 41618. * Epigenetic Noise (or methylation entropy) is the mechanistic increase in disorder, variability, and transcriptional drift across the entire epigenome, driven by incomplete DNA repair 102024. * Epigenetic Clocks are mathematical models and biomarkers that track specific, highly reproducible subsets of this age-related methylation drift to quantify biological age 1722.
While epigenetic clocks are highly accurate, ongoing debate questions whether the specific CpG sites measured by the clocks are the actual causal drivers of aging or merely passenger phenomena - reliable symptoms of the underlying entropic noise 2825.
The Backup Copy Hypothesis
A core tenet of the Information Theory of Aging is the assertion that the aging process is not an inevitable, one-way descent into irreversible decay. If aging is fundamentally an issue of corrupted information rather than destroyed physical hardware, the ITOA proposes that the process can theoretically be reversed 21821. However, for a cell to restore its youthful epigenetic landscape, it must possess a reference point. This necessitates the existence of a cellular "backup copy" - a retained, protected memory of the original youthful epigenome that survives the decades of accumulated noise 2232.
The exact physical and molecular nature of this backup copy remains one of the most actively investigated and debated aspects of the ITOA 26. Current models suggest that this information is shielded by a combination of "passive" and "active" observers within the cell's molecular architecture 26. Passive observers may include highly stable DNA modifications, specific topological domains of DNA rich in CpG islands, or robust protein complexes that act as physical barriers 26. For example, proteins like QSER1 have been identified as potential critical factors in retaining epigenetic information by protecting essential developmental genes from hypermethylation 214. Active observers, conversely, might interface directly with reprogramming machinery to mark regions requiring restoration, guiding epigenetic modifiers back to their youthful loci during a rejuvenation event 26.
Partial Epigenetic Reprogramming
The mechanism by which this hypothesized backup copy is accessed is known as epigenetic reprogramming. In 2006, Shinya Yamanaka revolutionized stem cell biology by demonstrating that the introduction of four specific nuclear transcription factors - OCT4, SOX2, KLF4, and c-MYC (collectively known as OSKM) - could completely erase the somatic identity of an adult cell. This process forces the cell back into an induced pluripotent stem cell (iPSC) state, effectively resetting its epigenetic age to zero 22728.
While full OSKM reprogramming is an invaluable tool in vitro, its continuous systemic application in vivo is highly toxic. Complete dedifferentiation strips cells of their specialized identities. If an animal is continually exposed to OSKM factors, its tissues undergo dysplasia, organs fail as cells lose their functional characteristics, and the subject rapidly develops complex tumors known as teratomas, leading to rapid mortality 15272930.
To harness the rejuvenating power of reprogramming safely, researchers pioneered the concept of partial or transient reprogramming 152930. By expressing the Yamanaka factors for short, carefully controlled, cyclical durations, researchers aim to decouple the rejuvenation of biological age from the dangerous loss of somatic identity 273038. Often, this involves omitting the oncogene c-MYC entirely to create a safer, three-factor "OSK" cocktail 2730.
Partial reprogramming gently pushes the cell back up the Waddington landscape - just far enough to erase the accumulated epigenetic noise and restore youthful chromatin configurations, but not far enough to cross the threshold into stem-cell pluripotency 82938. Evidence from multiple laboratories indicates that transient exposure successfully reactivates the theorized backup copy, allowing the cell to "remember" its original state, clear heterochromatic aberrations, and restore youthful gene expression patterns without forgetting its identity 22931.
| Feature | Full Reprogramming (OSKM) | Partial Reprogramming (OSK / Transient) |
|---|---|---|
| Factors Used | OCT4, SOX2, KLF4, c-MYC 27. | Typically OCT4, SOX2, KLF4 (c-MYC excluded due to oncogenic risk) 2730. |
| Duration of Expression | Continuous (weeks) 2729. | Transient, cyclical, or pulsed (days) 152930. |
| Cellular Identity | Completely erased; cell reverts to pluripotent stem cell (iPSC) 227. | Preserved; cell maintains original somatic lineage and function 273038. |
| In Vivo Outcome | Tissue dysplasia, teratoma formation, rapid mortality 152729. | Improved tissue function, restored vision, extended healthspan, age reversal 272932. |
| Epigenetic Clock Status | Reset to zero 215. | Significantly reduced to a more youthful biological age 152731. |
Experimental Validation: The ICE Mouse Model
To rigorously test whether epigenetic noise is a fundamental cause of aging rather than merely a downstream symptom of other degradative processes, David Sinclair's laboratory at Harvard Medical School developed the Inducible Changes to the Epigenome (ICE) mouse model 8213233. The objective of this landmark study was to isolate the variable of epigenetic disruption from the traditional paradigm of accumulating genetic sequence mutations.
The ICE model utilizes an engineered endonuclease, I-PpoI, derived from the slime mold Physarum polycephalum 21. This enzyme was integrated into the mouse genome under the control of a tamoxifen-inducible promoter 2133. When activated by the administration of tamoxifen, I-PpoI generates DNA double-strand breaks at roughly 20 specific, predictable sites across the mammalian genome.
Crucially, the I-PpoI enzyme generates "clean" cuts that the mammalian cellular machinery repairs with near-perfect fidelity. Extensive sequencing confirmed that the ICE system induces these specific breaks without increasing the overall background frequency of genetic mutations in the treated mice 82132. This allowed the researchers to uncouple DNA damage from DNA mutation.
By inducing these non-mutagenic DSBs for three weeks in young (4 - 6 month-old) mice, the researchers forced the Relocalized Chromatin Modifier pathway into overdrive 821. The cellular repair factors continuously mobilized, sealed the DNA flawlessly, and attempted to return to their native loci. However, the sheer volume of repair events generated a massive influx of epigenetic noise, simulating the epigenetic wear and tear of decades of life within a matter of weeks 82133.
The results were striking. By 10 months of age, the ICE-treated mice exhibited severe phenotypes indistinguishable from natural aging. They displayed physiological frailty, hair loss, reduced body mass, kyphosis, and cognitive decline 82132. At the molecular level, their cells exhibited widespread ex-differentiation, heterochromatin loss, and an advancement of the DNA methylation clock, rendering their tissues molecularly 1.5 times "older" than control mice 82132. Because the underlying DNA sequence was confirmed to be largely unmutated, the ICE study provided highly compelling evidence that the erosion of the epigenetic landscape is sufficient to drive the aging process independently of sequence mutations 2132.
To demonstrate that this process was reversible - a core requirement of the ITOA - the researchers subsequently delivered the OSK genes via an adeno-associated viral (AAV) vector to the artificially aged ICE mice, as well as to naturally aged mice 82732. The transient activation of OSK successfully counteracted the aging effects, decreasing the expression of age-associated markers in the kidneys and muscles 832. Most notably, the treatment restored youthful DNA methylation patterns in retinal ganglion cells, promoting axon regeneration after crush injury and functionally restoring vision in both a mouse model of glaucoma and in normal old mice 827.
Risks and Limitations of In Vivo Reprogramming
While the ITOA and the success of partial reprogramming offer a profound shift in gerontological biology, the translation of these techniques into safe, systemic clinical therapies faces substantial technical and biological hurdles 154234.
The primary risk remains the inherently stochastic and heterogeneous nature of the reprogramming process. Even brief or low-level systemic induction of pluripotency factors can inadvertently push a fraction of highly sensitive cells past the safety threshold into dedifferentiation, triggering the aforementioned teratoma formation 3042.
Achieving precise spatiotemporal control of OSK expression across a complex, multicellular organism is technically demanding. Systemic delivery via viral vectors or doxycycline-inducible systems often yields unequal induction. Organs characterized by naturally high cellular plasticity and rapid turnover, such as the liver and the intestine, absorb reprogramming factors quickly and are highly vulnerable to runaway dedifferentiation 4244. Continuous or excessive expression specifically in these tissues rapidly compromises normal organ function, leading to intestinal malabsorption, severe body weight loss, and premature death long before other tissues register rejuvenating benefits 384244.
Additionally, the localized tissue microenvironment plays a confounding role in reprogramming efficacy. Pro-inflammatory signals, which are heavily secreted by the senescent cells prevalent in aged tissues, can hyper-sensitize neighboring cells to the Yamanaka factors. This inflammatory context makes it difficult to predict safe dosing windows, as inflamed tissues may reprogram too rapidly and lose their identity 42. Consequently, early clinical trials (such as those exploring OSK therapies) are strictly limiting exposure to contained, immune-privileged environments like the eye, where viral delivery can be precisely controlled and off-target systemic effects minimized 42.
Academic Critiques and Competing Paradigms
The postulation of the Information Theory of Aging has sparked robust academic debate, challenging long-held dogmas and inviting rigorous scientific scrutiny. Critics have focused on potential methodological confounders in the foundational experiments, alternative interpretations of the "backup copy," and a resurgence of genomic data supporting somatic mutations as the true upstream cause of aging.
Methodological Critiques of the ICE Model
Prominent researchers, including Charles Brenner and James Timmons, have published formal critiques questioning the interpretations derived from the ICE model data 45. A primary concern centers on the physiological consequences of using the I-PpoI endonuclease to induce DNA double-strand breaks. Brenner points out that introducing widespread DSBs intrinsically triggers the p53 tumor suppressor pathway - a massive cellular stress response mechanism that naturally induces cell cycle arrest, apoptosis (cell death), and cellular senescence 204535.
Critics postulate that the accelerated aging phenotype observed in ICE mice may be heavily confounded by systemic p53-mediated tissue stress and cell death, rather than being a pure demonstration of epigenetic information loss driving natural aging 45. Furthermore, critics have noted a deficiency in broad functional data. While the original Sinclair studies highlight impressive molecular marker rejuvenation and specific visual restoration, skeptics argue that there is insufficient physiological data demonstrating that other complex, aged organs in the ICE mice were genuinely restored to youthful operational capacity following OSK treatment 45.
The Causality Debate: Mutations vs. Epigenetic Noise
The most profound theoretical challenge to the ITOA originates from high-resolution genomic studies, culminating in a pivotal 2025 study published in Nature Aging by Trey Ideker and colleagues 3637. The ITOA dictates that epigenetic noise drives aging downstream of DNA repair, and that somatic sequence mutations play a secondary, non-causative role 232. However, by analyzing genomes across thousands of patient tissue samples, Ideker's team demonstrated a highly predictable, causal link functioning in the opposite direction: permanent genetic mutations actively drive the epigenetic changes that characterize biological aging 374938.
This research provided a mechanistic link showing that somatic mutations directly dictate the ticking of the "epigenetic clock." When a CpG site in the DNA sequence mutates, or when an unmethylated cytosine spontaneously deaminates into a thymine, it creates an immediate local epigenetic disruption . The study revealed that a single point mutation causes a massive "ripple effect" or "explosion of methylation change," altering the epigenome in highly predictable patterns up to 10,000 nucleotide bases away from the original mutation site 37383940.
If somatic mutations are the fundamental, upstream driver of epigenetic drift - and epigenetic clocks are merely tracking the downstream fallout of irreversible genetic damage - then the implications for anti-aging therapies are severe. Attempting to cure aging solely by resetting the epigenetic software via partial reprogramming may only address the downstream symptoms, leaving the underlying mutated hardware unchanged and vulnerable to immediate re-corruption 36373839.
| Feature | Somatic Mutation Theory | Information Theory of Aging (ITOA) |
|---|---|---|
| Primary Driver of Aging | Accumulation of irreversible DNA sequence mutations over time 374142. | Loss of reversible epigenetic information and cellular identity (epigenetic noise) 12. |
| Role of Epigenetics | Epigenetic changes (methylation drift) are a downstream symptom or consequence of underlying mutations 3840. | Epigenetic noise is the upstream, primary cause of physiological decline 221. |
| Reversibility of Aging | Fundamentally irreversible without complex, currently unfeasible genome editing to fix mutated sequences 3742. | Highly reversible by accessing a cellular "backup copy" via partial epigenetic reprogramming 12. |
| Primary Evidence Base | Mutation burden scales inversely with species lifespan; point mutations trigger massive epigenetic ripple effects 40425543. | ICE mice show aging with non-mutagenic DSBs; OSK factors reverse epigenetic age and restore functional capabilities 212732. |
The Double Code Hypothesis
Another rigorous critique of Sinclair's framework is articulated in the recently formulated "Double Code Hypothesis of Ageing" (emerging in literature across 2025 - 2026) 325744. This model accepts that epigenetic information loss is a central feature of aging, but it sharply criticizes the ITOA for relying on a mysterious, undefined "backup copy" supposedly hidden somewhere within the adult somatic cell 265744.
The Double Code Hypothesis argues that Sinclair's framework possesses a fundamental conceptual flaw: it neglects the reality of epigenetic intergenerational inheritance 2657. According to this alternative model, the "backup copy" of youthful epigenetic information is not an abstract observer waiting in an adult skin or liver cell. Rather, the true backup copy is the pristine epigenome inherited directly from the parents, which is selectively protected, restored, and reset only during the highly specialized process of meiosis (the creation of germ cells) 57.
Under this framework, biological information relies on a dual inheritance system - both the stable genetic genome and the highly plastic epigenome must remain functionally aligned 57. Aging is defined as the inevitable, progressive loss of coordination between these two layers during mitotic division in somatic cells. The Double Code Hypothesis suggests that aging is not merely an software error waiting to be corrected by cellular reprogramming, but rather an inherent instability built into complex multicellular organisms - an evolutionary side consequence of how life transmits both stable and adaptive codes across generations 3257.
Conclusion
The Information Theory of Aging represents a paradigm-shifting lens through which to view the biological deterioration of complex life. By reframing aging not merely as the irreversible decay of physical "hardware" (mutations) but as the corruption of dynamic "software" (epigenetic information), the ITOA provides a highly cohesive mechanistic explanation for how diverse environmental stressors can collectively erode cellular identity. Through the Relocalized Chromatin Modifier hypothesis, the model traces how emergency DNA repair responses inadvertently scatter vital silencing proteins, leaving behind a persistent residue of epigenetic noise that ultimately drives heterochromatin loss, inflammaging, and cellular ex-differentiation.
The most compelling, yet highly debated, component of the ITOA is its optimistic assertion of reversibility. The concept that somatic cells harbor a persistent "backup copy" of their youthful epigenetic state - accessible via the transient expression of Yamanaka factors - has moved age-reversal from theoretical speculation to laboratory reality, as evidenced by in vivo models demonstrating the restoration of vision and tissue function.
However, the field remains deeply contested. Major clinical risks regarding teratoma formation, organ-specific toxicity, and runaway cellular dedifferentiation must be rigorously overcome before epigenetic reprogramming can be safely utilized systemically. Furthermore, profound academic critiques persist regarding the fundamental directionality of causality. Emerging high-resolution genomics suggest that somatic sequence mutations may indeed instigate the very epigenetic clock drift that the ITOA blames for aging, implying that resetting the epigenetic software may ultimately prove insufficient if the underlying genetic hardware remains irrevocably damaged. Meanwhile, alternative evolutionary frameworks like the Double Code Hypothesis suggest that the true reset of epigenetic information belongs strictly to the domain of intergenerational inheritance. Navigating the friction between these competing theories will define the next decade of gerontology, determining whether aging can be broadly cured via a localized epigenetic reset, or if it remains an intrinsic, multifaceted limitation of biological complexity.