Impact of prenatal conditions on lifelong health
The Developmental Origins of Health and Disease (DOHaD) hypothesis represents a paradigm shift in the understanding of human biology, proposing that environmental exposures during critical periods of early development - spanning preconception, fetal growth, and early infancy - permanently shape the physiological and metabolic trajectory of the organism 123. While evolutionary biology has long recognized that developmental plasticity allows organisms to adapt to immediate environmental constraints, the DOHaD framework demonstrates that these early biological adaptations frequently dictate susceptibility to non-communicable diseases (NCDs) in adulthood, including cardiovascular disease, type 2 diabetes, metabolic syndrome, and psychiatric disorders 245.
By integrating insights from epidemiology, epigenomics, and environmental health, life-course epidemiology has established that human health is not merely a product of adult lifestyle choices interacting with a static genetic code 678. Instead, it is an evolving biological ledger of the ancestral and early-life environments 69. The mechanisms underlying these phenomena extend beyond mere nutritional deficits, encompassing environmental toxicants, microbiome assembly, maternal immune activation, structural social determinants, and paternal epigenetics 1101112.
Foundational Frameworks and Hypotheses
The modern iteration of DOHaD originated in the late 1980s when epidemiologist David Barker identified a robust inverse correlation between low birth weight and the subsequent risk of ischemic heart disease and metabolic dysfunction in adulthood 24. Initially termed the "Barker hypothesis," the scope of the field has expanded significantly, recognizing that birth weight is merely a crude, proxy indicator of the intrauterine environment, and that profound fetal programming can occur even when birth weight falls within normal parameters 1314.
The Thrifty Phenotype Hypothesis
To explain the epidemiological correlations between low birth weight and adult disease, researchers formulated the "thrifty phenotype" hypothesis. This concept posits that when a fetus experiences inadequate maternal nutrition or placental insufficiency, it enacts a triage mechanism, prioritizing the allocation of scarce nutrients to essential organs - most notably the brain - at the expense of other tissues such as the endocrine pancreas, skeletal muscle, and kidneys 151617.
This adaptive diversion ensures short-term fetal survival but permanently alters the architectural endowment and functional capacity of the neglected organs. For example, a reduction in the total number of nephrons or pancreatic beta cells limits the organ's functional reserve, rendering the individual highly susceptible to hypertension or insulin resistance when challenged by age or caloric excess in adulthood 161718.
The Predictive Adaptive Response
The thrifty phenotype concept was later expanded into the Predictive Adaptive Response (PAR) framework, primarily developed by Peter Gluckman and Mark Hanson 1317. The PAR model proposes that the developing organism utilizes early-life environmental cues to forecast the conditions of its future postnatal environment, adjusting its developmental trajectory accordingly 131519. If a fetus anticipates a nutrient-poor environment, it alters its physiological set-points to maximize energy conservation and fat storage 1919.
A biological "mismatch" occurs when the forecasted environment differs significantly from the actual adult environment 1319. Disease risk increases exponentially when a fetus programmed for scarcity is born into a modern, obesogenic environment characterized by caloric abundance, leading to rapid postnatal catch-up growth and severe metabolic strain 171920.
Mathematical modeling of evolutionary health underscores the distinction between PARs and basic developmental constraints. Developmental constraint (DC) models predict that a low-quality developmental environment uniformly produces poor adult outcomes 21. Conversely, PAR models emphasize that outcomes depend on the interaction between early and late environments. Statistical simulations demonstrate that PARs are more accurately detected using quadratic regression models rather than standard linear interaction models, highlighting that physiological buffering operates effectively within a specific, matching environmental range before leading to pathology 21.
Integration with Neurodevelopmental Critical Periods
Recent scholarship increasingly integrates the DOHaD paradigm with the framework of sensitive and critical periods in neurobiology 22. During early development, the central nervous system exhibits heightened plasticity, allowing the brain's circuitry to be permanently sculpted by sensory and environmental inputs 2223. Prenatal insults - ranging from maternal psychosocial stress to substance exposure - can alter the trajectory of these critical periods. When DOHaD adaptations alter the timing or duration of these windows of neuroplasticity, it affects the offspring's subsequent capacity to learn from and adapt to the postnatal environment, thereby embedding long-term vulnerabilities to cognitive deficits and mood disorders 2223.
Epidemiological Evidence from Famine Cohorts
Historical famines have served as tragic but invaluable natural experiments for DOHaD research, providing unique insights into the critical windows of fetal vulnerability. However, the interpretation of these retrospective cohorts requires rigorous methodological scrutiny, as recent analytical advances have challenged long-standing assumptions.
The Dutch Hunger Winter
The Dutch Hunger Winter (1944 - 1945) remains one of the most rigorously studied cohorts because the famine was strictly delineated in time, allowing researchers to isolate the effects of severe maternal undernutrition (400 - 800 calories per day) based on the specific trimester of gestational exposure 1520. The data reveal that the timing of exposure strictly dictates the resulting phenotype, underscoring the temporal specificity of developmental programming 20.
| Trimester of Exposure | Immediate Fetal Impact | Long-Term Adult Phenotype |
|---|---|---|
| Early Gestation | Normal birth weight. | Higher rates of adult obesity, elevated cardiovascular disease risk, altered lipid profiles, and impairment in selective cognitive attention. |
| Mid-Gestation | Reduced birth weight. | Reduced renal function markers, increased risk of obstructive airway disease, and metabolic disturbances. |
| Late Gestation | Reduced birth weight (small for gestational age). | Maintained smaller body size throughout life; paradoxically exhibited lower rates of adult obesity compared to individuals exposed during early gestation. |
Table 1: Differential effects of severe nutrient deprivation timing during the Dutch Hunger Winter on human health outcomes 20.
Individuals exposed during early gestation experienced profound central nervous system and metabolic programming, yet were born with normal birth weights, reinforcing the principle that birth weight is an insufficient proxy for fetal programming 20. Because post-war prosperity returned quickly to the Netherlands, these individuals experienced a severe PAR mismatch, suffering high rates of metabolic disease. In contrast, populations exposed to the prolonged Siege of Leningrad, where postwar shortages persisted, did not exhibit the same spike in obesity, further validating the mismatch hypothesis 20.
The Chinese Great Famine and Methodological Debates
The Chinese Great Famine (1959 - 1961), responsible for an estimated 30 million excess deaths, resulted from systemic policy failures, agricultural restructuring, and central planning inflexibility 242526. For decades, studies of famine survivors indicated that prenatal exposure drastically increased the risk of type 2 diabetes, hypertension, and metabolic syndrome 27.
However, systematic re-analyses have highlighted significant methodological vulnerabilities in these conclusions. Researchers at Columbia University re-evaluated previous cohorts, identifying an unrecognized age-cohort flaw: numerous earlier studies compared famine births strictly to control groups born after the famine 27. Because the famine cohort was intrinsically older than the control cohort, the natural accumulation of age-related diseases artificially inflated the apparent metabolic effects of the famine 27.
When the researchers neutralized this age effect by integrating controls born before the famine, the reported increases in diabetes, high blood pressure, and related chronic conditions vanished 27. The only chronic condition that remained significantly elevated among the famine-exposed cohort was schizophrenia. This recalibration suggests that severe early-life starvation may uniquely and permanently disrupt neurodevelopmental architecture, but that generalized metabolic programming may be more nuanced or population-dependent than previously assumed 27.
Molecular Mechanisms of Fetal Programming
The translation of environmental exposures into stable, lifelong phenotypic traits is governed by molecular mechanisms that alter cellular function without changing the underlying genetic sequence. The primary biological substrate for these adaptations resides in the epigenome, the mitochondrion, and telomere biology 117.
Epigenetic Modifications and Chromatin Remodeling
Epigenetic regulation controls gene expression through chemical modifications to DNA and structural alterations to chromatin 1728. During embryogenesis, the epigenome undergoes comprehensive reprogramming to establish pluripotency, followed by rapid, lineage-specific re-establishment of epigenetic marks 2930. Environmental stimuli during this highly plastic phase can permanently skew the trajectory of cellular differentiation 129. The primary mechanisms include:
- DNA Methylation: The covalent addition of a methyl group to cytosine residues, typically at cytidine-guanosine (CpG) sequences within promoter regions. Hypermethylation generally induces a transcriptionally inactive chromatin state, suppressing gene expression 1731.
- Histone Modifications: Post-translational alterations (such as acetylation, methylation, phosphorylation, and sumoylation) to the histone proteins around which DNA is coiled. Histone acetylation typically decreases the binding affinity between histones and DNA, creating an "open" euchromatin structure that facilitates active transcription, whereas specific methylation marks (e.g., H3K9me3, H3K27me3) induce chromatin condensation and gene repression 173032.
- Non-coding RNAs (ncRNAs): MicroRNAs (miRNAs) and long non-coding RNAs regulate gene expression post-transcriptionally by binding to target messenger RNAs, either promoting their degradation or inhibiting their translation into proteins 1017.
Recent synthesis of DOHaD and gerontology proposes the "Pathological Epigenetic Events that are Reversible" (PEER) hypothesis, which posits significant overlap between the hallmarks of aging and the hallmarks of early-life developmental programming 33. Exposures to environmental toxicants or severe maternal stress induce cellular senescence and epigenetic alterations that artificially accelerate biological age, though emerging models suggest these overlapping pathways may be partially reversible through targeted metabolic interventions 3334.
Telomere Attrition and Cellular Senescence
Telomeres are nucleoprotein caps at the ends of chromosomes that protect genomic integrity. Telomere length (TL) progressively shortens with cell division, serving as an internal clock for cellular aging 3537. Accelerated telomere attrition is associated with premature cellular senescence, psychiatric disorders, and early mortality 3536.
The fetal programming of telomere biology hypothesis outlines how the intrauterine environment calibrates the initial setting of offspring TL 35. Maternal exposure to psychological stress, adverse childhood experiences (ACEs), and chronic illness elevates systemic inflammation and maternal cortisol levels, which accelerate oxidative stress and impair telomerase activity in the developing fetus 3537. Longitudinal studies examining mother-child dyads reveal that infants exposed to relapsing maternal depressive symptoms exhibit significantly shortened buccal telomere lengths by age 3, independently of other demographic confounders 3537.
Furthermore, shortened fetal telomeres are intimately linked to abnormal organogenesis. Analyses of amniotic fluid indicate that TL is significantly reduced in fetuses exhibiting developmental anomalies, with maternal age-adjusted TL explaining up to 38% of the variance in fetal TL 38. Metabolic disturbances such as gestational diabetes mellitus (GDM) further compromise the intrauterine environment, increasing oxidative stress and resulting in significant telomere attrition in the umbilical cord blood mononuclear cells of the offspring 36.
Mitochondrial Dysfunction and Oxidative Stress
Mitochondria govern cellular energy metabolism, oxidative phosphorylation, and the regulation of reactive oxygen species (ROS) 1839. Because the mitochondrial genome (mtDNA) is highly vulnerable to oxidative damage, intrauterine growth restriction (IUGR) and maternal malnutrition can induce persistent mitochondrial dysfunction 1839.
In models of IUGR, researchers have documented paradoxical increases in fetal cardiac mtDNA content and respiratory chain proteins - likely an initial, maladaptive compensatory response to nutrient restriction 18. Despite increased mitochondrial mass, overall metabolic function remains fundamentally compromised, characterized by impaired complex I and II/III activities 18. High levels of mitochondrial ROS during the pre-implantation and embryonic stages actively disrupt the telomere elongation process that normally occurs at the morula-to-blastocyst transition, creating a direct mechanistic link between maternal metabolic health, mitochondrial efficiency, and the cellular aging clock of the offspring 37.
Neuroendocrine Alterations and Maternal Stress
The biological embedding of maternal stress is primarily mediated through the maternal-placental-fetal neuroendocrine interface, specifically the hypothalamic-pituitary-adrenal (HPA) axis 1723.
The Hypothalamic-Pituitary-Adrenal Axis
Under standard physiological conditions, the fetus is protected from high concentrations of maternal circulating cortisol by the placental enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), which converts active cortisol into inactive corticosterone 4041. However, severe maternal psychosocial stress, hypoxia, or the ingestion of 11β-HSD2 inhibitors (such as licorice) can overwhelm or bypass this enzymatic barrier, leading to fetal glucocorticoid overexposure 4041.
Clinically, synthetic glucocorticoids are routinely administered to pregnant women presenting with threatened preterm labor to accelerate fetal lung maturation. While this intervention is critical for acute neonatal survival, it fundamentally mimics a state of severe physiological stress and has been linked to reduced birth weight and restricted head circumference 4042.
Long-Term Cardiovascular and Psychiatric Outcomes
Elevated fetal glucocorticoids trigger premature terminal differentiation of specific cell populations, notably limiting the proliferation of cardiomyocytes and renal nephrons 161841. A reduced structural endowment of these critical functional units at birth inherently limits the organ's physiological reserve, predisposing the offspring to hypertension, pathological cardiac remodeling, and cardiovascular failure in adulthood 161841.
Furthermore, glucocorticoid overexposure structurally and functionally remodels the developing fetal brain 23. Prolonged elevations in cortisol alter the set-point and feedback sensitivity of the offspring's own HPA axis. This permanent dysregulation manifests postnatally as altered stress reactivity, heightening the individual's lifelong vulnerability to mood disorders, anxiety, depression, and generalized psychopathology 234043.
The Maternal-Infant Microbiome Axis
Emerging research situates the maternal-infant microbiome axis as a central regulatory network within the DOHaD paradigm. This axis represents a dynamic interface linking maternal microbial niches with neonatal immune and metabolic development through the transfer of microbial consortia, metabolites, and epigenetic signals 104445.
Early Microbial Succession and Global Patterns
The first 1,000 days of life define a critical window for the assembly of the infant gut microbiome, transitioning from a near-sterile fetal environment to a complex, mature microbial community 4647. Extensive cohort analyses spanning populations across multiple continents have revealed that, despite profound geographical and socio-economic differences, infants universally exhibit similar patterns of microbial succession 47.
Early infancy is heavily dominated by Bifidobacterium species, which specialize in metabolizing human milk oligosaccharides 4547. Upon weaning and the introduction of solid foods, this dominance recedes in favor of Faecalibacterium prausnitzii and Lachnospiraceae species, accompanied by a predictable increase in microbial alpha diversity 47. Disruptions to this highly conserved sequence - driven by maternal antibiotic administration, cesarean delivery, or suboptimal neonatal feeding practices - deprive the developing immune system of necessary microbial stimuli. This "microbial dysbiosis" interrupts the induction of immune tolerance, leaving the offspring predisposed to inappropriate inflammatory responses, atopic eczema, asthma, and inflammatory bowel diseases later in life 444547.
Maternal Immune Activation (MIA)
The concept of Maternal Immune Activation (MIA) aligns closely with DOHaD theory by providing a mechanistic explanation for how maternal infections disrupt fetal development 48. In MIA models, pregnant subjects are exposed to immune-activating agents (such as poly I:C, which mimics a viral infection, or lipopolysaccharide, which mimics a bacterial infection). The resulting maternal cytokine storm directly impacts fetal hematopoiesis and alters the development of the fetal immune system 48. Beyond increasing the risk of neurodevelopmental disorders, MIA alters the programming of the offspring's bone marrow and pulmonary immune cell compartments, rendering the adult offspring highly susceptible to allergic asthma and immune dysregulation 48.
Epigenetic Imprinting via Microbial Metabolites
The maternal microbiome directly influences the fetal epigenome through the transfer of microbially derived metabolites 1049. Maternal gut bacteria ferment dietary fibers to produce short-chain fatty acids (SCFAs), primarily butyrate, acetate, and propionate. These SCFAs cross the placenta and function as potent, natural histone deacetylase (HDAC) inhibitors 10.
By inhibiting HDACs, maternally derived butyrate enhances histone acetylation at critical immune-regulatory genetic loci within the fetus, facilitating the differentiation and maturation of fetal T-regulatory cells 10. Additionally, the maternal microbiome contributes essential one-carbon donors (such as folate, choline, and betaine) required for DNA methylation. Maternal dysbiosis restricts the supply of these critical metabolites, leading to the aberrant hypermethylation of key metabolic genes, such as leptin, thereby predisposing the offspring to impaired energy regulation and obesity 1049.
Environmental Toxicants and Endocrine Disruption
While DOHaD research initially centered on maternal nutrition, the field has aggressively expanded to assess the multi-generational impacts of environmental chemical exposures 1150. Endocrine-disrupting chemicals (EDCs) readily cross the placental barrier, interfering with fetal hormonal signaling and developmental programming 1151.
Bisphenols, Phthalates, and Sex-Specific Vulnerabilities
EDCs are ubiquitous synthetic compounds utilized in the manufacture of plastics, food packaging, and personal care products 5255. Under normal physiological conditions, the fetus is protected from high concentrations of maternal estrogens by plasma binding proteins such as $\alpha$-fetoprotein. However, synthetic EDCs often possess a low binding affinity for $\alpha$-fetoprotein, allowing them to bypass this defense mechanism and directly engage fetal endocrine receptors 53.
| Chemical Class | Primary Environmental Sources | Observed Long-Term Offspring Health Impacts |
|---|---|---|
| Bisphenol A (BPA) | Polycarbonate plastics, epoxy resin can linings, thermal receipt paper. | Altered metabolic homeostasis, increased adiposity, disrupted mammary gland development, elevated risk of early puberty, and increased likelihood of persistent asthma specifically in males. |
| Phthalates (e.g., DEHP, MnBP) | Plasticizers in PVC, cosmetics, fragrances, and medical tubing. | Anti-androgenic effects, reduced anogenital distance in male offspring, impaired glucose tolerance, and increased incidence of childhood anxiety and sleep disorders. |
| Per- and Polyfluoroalkyl Substances (PFAS) | Water-resistant fabrics, non-stick cookware, firefighting foams. | Altered lipid metabolism, immunosuppression, and poorer overall metabolic syndrome scores in childhood, with more pronounced metabolic disruptions frequently observed in female offspring. |
Table 2: Health impacts of prenatal exposure to common Endocrine-Disrupting Chemicals (EDCs) 525553545556.
Prenatal exposure to complex EDC mixtures has been shown to degrade placental vasculature and alter steroidogenesis, suppressing both maternal estradiol and progesterone production 5556. Multi-omics studies from international cohorts demonstrate that these exposures exert pronounced, sex-specific effects on the offspring. For instance, high prenatal BPA exposure is closely associated with persistent asthma and metabolic syndrome predominantly in male offspring, while specific PFAS and PCB mixtures exert stronger metabolic disruptions in females 525457.
Structural Determinants and Climate Vulnerability
The biological phenomena documented by DOHaD research do not occur in a vacuum; they operate within the macro-environment mapped by the Social Determinants of Health (SDOH) 585960. The biology that allows human fetuses to adapt and survive works against marginalized populations when they are persistently confronted with the toxic stress of poverty, systemic inequality, and inadequate healthcare systems 5860.
Social Determinants and Reframing Maternal Blame
Poverty, structural racism, food insecurity, and poor housing quality dictate the baseline nutritional and stress profiles of pregnant individuals 586061. Furthermore, hospital-level factors - including poor care coordination, staffing shortages, and discriminatory organizational practices - directly contribute to persistent racial and ethnic disparities in severe maternal morbidity and adverse neonatal outcomes 62.
As DOHaD findings permeate public health discourse, scholars warn against the unintended consequence of "maternal blame." The intense focus on the maternal-fetal dyad in both media and scientific literature creates a feedback loop that holds mothers almost uniquely culpable for their offspring's lifelong disease risk, often ignoring the upstream social, economic, and environmental factors that govern maternal behavior 963. Public health communication must shift from individual-level blame to identifying the structural systems that expose pregnant individuals to DOHaD-related risks in the first place 963.
Climate Change Impacts on Developmental Origins
Climate change acts as a profound multiplier of early-life adversity. Data aggregated by The Lancet Countdown underscores that extreme weather events, prolonged droughts, and increasing heatwaves severely threaten maternal food security and healthcare infrastructure globally 646566. High ambient temperatures and exposure to wildfire particulate matter (PM2.5) during gestation are increasingly correlated with disruptions in fetal neurodevelopment, intrauterine growth restriction, and adverse mental health outcomes in childhood 6467. The compounding burdens of environmental degradation and socioeconomic marginalization dictate that the earliest biological imprints of climate change are borne disproportionately by the world's most vulnerable populations 596667.
Paternal Contributions to Offspring Health
Historically, the assumption of "causal primacy" regarding maternal pregnancy effects resulted in severe data gaps regarding the father's role in developmental programming 6368. To rectify this imbalance, the field has formalized the study of the Paternal Origins of Health and Disease (POHaD), emphasizing that male-mediated developmental toxicity plays a substantial role in offspring health trajectories 126970.
Sperm Epigenetics and Preconception Health
Spermatozoa deliver far more than a haploid genome; they transmit a complex profile of epigenetic information, including DNA methylation marks, histone retention profiles, and small non-coding RNAs (sncRNAs) that guide early embryonic development 7172.
Paternal lifestyle factors during the preconception period dictate this epigenetic landscape. Paternal undernutrition, excessive saturated fat intake, and elevated Body Mass Index (BMI) drastically alter the methylation signatures of sperm 61272. In experimental models, paternal high-fat diets reliably induce metabolic syndrome, glucose intolerance, and increased adiposity in the F1 generation, completely independent of the maternal intrauterine environment 127072. Similarly, paternal exposure to environmental toxicants, radiation, and tobacco smoke prior to conception leads to sperm DNA damage and altered ncRNA expression, raising the risk of neurodevelopmental abnormalities and respiratory conditions in the offspring 7072. Consequently, public health strategies must aggressively pivot toward integrated preconception healthcare that actively incorporates paternal lifestyle modifications 697071.
Transgenerational Epigenetic Inheritance
One of the most profound, yet fiercely debated, extensions of DOHaD is the concept of Transgenerational Epigenetic Inheritance (TEI) - the transmission of environmentally induced phenotypes across multiple generations without any corresponding alteration to the underlying DNA sequence 327374.
Distinguishing Intergenerational from Transgenerational Transmission
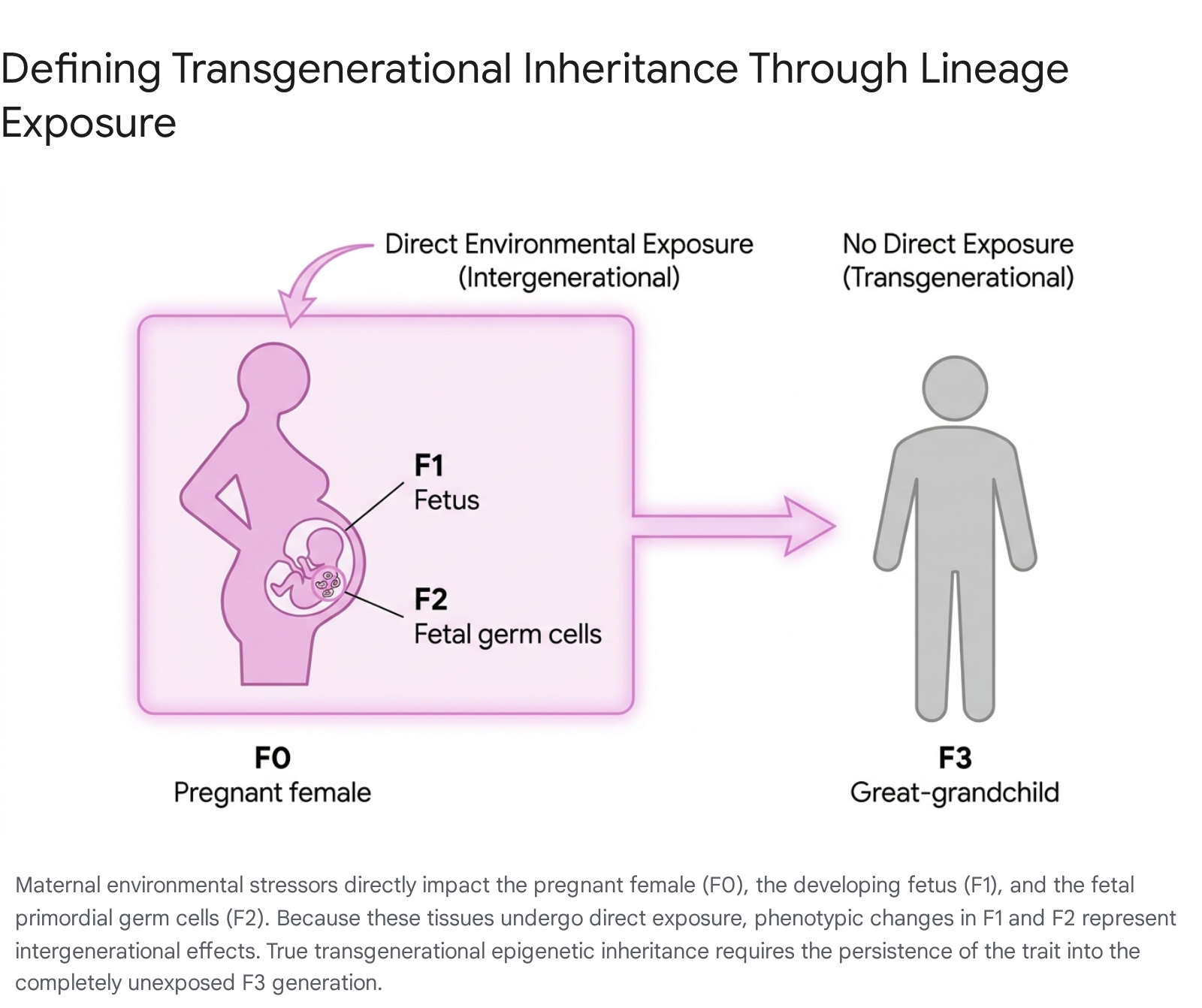
To accurately evaluate TEI, researchers must define exposure based on strict generational tracking (F0, F1, F2, F3) 3073. If a pregnant female (the F0 generation) is exposed to an environmental stressor such as a toxicant or famine, the developing fetus (the F1 generation) is directly exposed in utero. Concurrently, the primordial germ cells developing within that fetus - which will eventually mature into the eggs or sperm that create the F2 generation - are also directly exposed to the stressor 3073.
Therefore, any phenotypic changes observed in the F1 and F2 generations resulting from a maternal exposure are classified strictly as intergenerational inheritance, because the biological material that formed them had direct contact with the original environment 3073. True transgenerational epigenetic inheritance in a maternal lineage can only be confirmed if the phenotype persists into the F3 generation (the great-grandchildren), representing the first generation whose biology was never directly exposed to the ancestral stressor 3073. (For paternal exposures, where the F0 male is exposed, the F1 generation and their germ cells are exposed, making the F2 generation the first truly unexposed transgenerational cohort 5373).
Mammalian Germline Reprogramming
The existence of TEI in plants, nematodes, and Drosophila is well-documented, with specific histone methylation marks (e.g., H3K4me3, H3K9me3) known to pass intact across dozens of generations 3032. In mammals, however, the concept remains highly controversial and fiercely debated 3074.
Mammalian biology features two distinct waves of comprehensive epigenetic reprogramming - one immediately after fertilization and another during the specification of primordial germ cells - designed specifically to erase acquired environmental marks and restore cellular pluripotency 3074. For an environmentally induced epigenetic mark to be transmitted transgenerationally, it must successfully evade both waves of erasure 3074. While specific animal models have demonstrated multi-generational phenotypes arising from toxicant exposure, human evidence remains largely observational and strictly intergenerational, hampered by the long lifespan of humans and the near-impossibility of controlling for continuous, shared environmental and socio-economic exposures across multiple decades 303174.
Public Health Policy and Global Cohort Interventions
Translating the intricate molecular and physiological insights of DOHaD into actionable public health policies requires robust, long-term epidemiological data spanning diverse populations. To achieve this, researchers established the Consortium of Health-Orientated Research in Transitioning Societies (COHORTS), pooling massive data arrays from five major prospective birth cohorts in low- and middle-income countries: Pelotas (Brazil), INCAP (Guatemala), New Delhi (India), Cebu (Philippines), and Birth-to-Twenty (South Africa) 7576.
Following thousands of participants from birth well into adulthood, the COHORTS collaboration has provided definitive, population-level evidence that early-life nutrition and fetal growth trajectories strictly dictate adult human capital, cognitive attainment, and cardiovascular risk profiles 7576. The data uniformly underscores the necessity of moving public health interventions "upstream." Interventions targeting adult lifestyle modifications often yield minimal success in curbing NCDs because the physiological architecture of the disease was programmed decades earlier 77778.
Life Course Epidemiology and Adolescent Health Literacy
Integrating DOHaD into actionable policy requires a fundamental shift in healthcare resource allocation, prioritizing maternal and paternal preconception health, aggressively mitigating environmental toxicant exposure, and ensuring nutritional security during the first 1,000 days of life 71277.
Furthermore, educational interventions targeting adolescents have been identified as a highly effective mechanism for breaking the intergenerational cycle of disease. Programs designed to increase adolescent health literacy regarding DOHaD concepts empower young people to recognize how their current dietary and environmental exposures impact not only their own long-term health but the biological starting point of their future offspring 779. Ultimately, the DOHaD framework forces a comprehensive reevaluation of chronic disease etiology, proving that human health is not merely the product of adult choices and rigid genetics, but an evolving biological continuum shaped by the environments experienced across generations.