Impact of AlphaFold on protein folding and biological research
Historical Context of the Protein Folding Problem
Theoretical Foundations and Early Computation
The relationship between a protein's primary amino acid sequence and its functional, three-dimensional topological structure has served as a central paradigm in molecular biology for over six decades. The formalization of this relationship began with Christian Anfinsen's Nobel Prize-winning work in 1972, which demonstrated that denatured proteins spontaneously refold into their native conformations. This indicated that a protein's three-dimensional structure is thermodynamically determined by its primary sequence alone, establishing a 50-year-old grand challenge in computational biology: predicting a protein's structure strictly from its genetic code 1.
Early theoretical work highlighted the immense difficulty of this task. Cyrus Levinthal famously articulated what became known as Levinthal's Paradox, observing that a relatively simple 100-amino-acid protein theoretically possesses approximately $10^{47}$ possible spatial conformations 1. If a polypeptide chain were to sample all possible configurations randomly to find its energy minimum, it would require a timeframe exceeding the age of the universe to reach its native state. Therefore, protein folding must occur through highly biased, cooperative thermodynamic pathways rather than stochastic searching 1.
Initial computational attempts in the 1970s, such as the Chou-Fasman method, focused predominantly on predicting localized secondary structures, such as $\alpha$-helices and $\beta$-sheets, based on the statistical propensities of specific amino acids. However, these methods largely failed to predict tertiary interactions, yielding exceptionally low accuracy for global topologies 1. The field sought to quantify progress through the establishment of the Critical Assessment of protein Structure Prediction (CASP) in 1994, a biannual blind experiment designed to benchmark computational methods against unreleased, experimentally determined structures 1. Prior to 2018, the most successful computational methodologies relied heavily on template-based modeling (homology modeling), which required an existing homologous structure in the Protein Data Bank (PDB) to serve as a comparative scaffold 2. For ab initio targets lacking known structural templates, researchers utilized co-evolutionary analysis derived from multiple sequence alignments (MSAs) to predict contacting residues, combined with computationally expensive molecular dynamics simulations. Until CASP12 in 2016, contact prediction accuracy remained frustratingly low, plateauing at roughly 45% 1.
Traditional Experimental Methodologies
Before the advent of high-accuracy artificial intelligence models, structural biology relied exclusively on empirical biophysical techniques. The three dominant experimental methodologies - X-ray crystallography, Nuclear Magnetic Resonance (NMR) spectroscopy, and Cryo-Electron Microscopy (Cryo-EM) - each possess distinct mechanical principles, advantages, and constraints.
Historically acting as the gold standard of structural biology, X-ray crystallography accounts for approximately 84% of all structures deposited in the Protein Data Bank as of late 2024, with over 9,600 structures resolved by this method in 2023 alone 34. The technique involves coaxing highly purified proteins into forming well-ordered, solid-state crystals. These crystals are subsequently exposed to high-energy X-ray beams, often generated by highly specialized, multi-million-dollar third-generation synchrotrons. The periodic lattice of the protein crystal diffracts the X-rays, producing a pattern of Bragg reflections on a detector. Once the complex "phase problem" is mathematically resolved, researchers calculate electron density maps to build highly precise atomic models 356. While capable of unsurpassed atomic and sub-atomic resolution, the primary bottleneck of X-ray crystallography is the crystallization process itself. The methodology requires extensive optimization of pH, temperature, and precipitating agents, and many vital biological targets - particularly highly dynamic membrane proteins, intermediary structural states, and large multi-subunit molecular complexes - resist forming the necessary ordered lattices 356.
Nuclear Magnetic Resonance (NMR) spectroscopy provides entirely different molecular insights by analyzing biological macromolecules in an aqueous solution, a state that closely mimics their native physiological environments 37. NMR exploits the magnetic properties of atomic nuclei, measuring how they are perturbed by intra- and intermolecular interactions. This makes NMR uniquely valuable for extracting dynamic structural information, tracking conformational mobility, and studying intrinsically disordered protein regions 37. Despite these advantages, NMR has contributed to less than 10% of total PDB structures 4. The technique is severely limited by molecular weight constraints, generally proving ineffective for complex assemblies exceeding 40 to 50 kDa due to overlapping, uninterpretable spectral data 78. Furthermore, NMR requires highly concentrated, stable samples and relies on exceptionally expensive de-novo protein synthesis using isotopic labels such as $^{15}$N, $^{13}$C, and deuterium ($^2$H) 3.
Cryo-Electron Microscopy (Cryo-EM) emerged as a transformative technology designed to bypass the crystallization bottlenecks of X-ray diffraction. In Cryo-EM, purified samples are applied to a grid and flash-frozen in liquid ethane, trapping the molecules in a thin layer of non-crystalline vitreous ice 39. A transmission electron microscope is then used to capture thousands of two-dimensional projections of the molecules in various random orientations, which are computationally reconstructed into a three-dimensional density map 3. Following the so-called "resolution revolution" beginning around 2013 - driven by the invention of direct electron detectors (DEDs) that bypass mechanical shutters to read electrons at high frame rates, alongside advanced motion-correction algorithms - Cryo-EM now routinely rivals X-ray crystallography in resolution capabilities 347. It is uniquely suited for massive molecular machines, virions, and membrane proteins exceeding 250 kDa 79. By 2023 and 2024, Cryo-EM accounted for up to 40% of all new structure deposits 4. However, the technique demands extremely costly microscopy hardware, generates massive computational overhead for image classification, and generally struggles to resolve very small proteins 79.
| Analytical Parameter | X-ray Crystallography | Nuclear Magnetic Resonance (NMR) | Cryo-Electron Microscopy (Cryo-EM) |
|---|---|---|---|
| Physical State | Static crystalline solid | Aqueous solution | Vitrified non-crystalline ice |
| Primary Advantage | Unsurpassed atomic precision; rapid model building | Captures solution dynamics and flexible disordered regions | No crystallization required; captures multiple native states |
| Primary Limitation | Heavy reliance on unpredictable crystallization | Restricted to low molecular weights; expensive isotopes | Highly expensive hardware; computationally intensive data processing |
| Molecular Weight Range | Broad range, but massive complexes resist crystallization | Strictly limited (typically < 40-50 kDa) | Ideal for large macromolecules (> 250 kDa) |
| PDB Contribution (2023-2024) | Dominant historical method (~66-84% of total repository) | Minor contribution (~1.9% to 10% annually) | Rapidly ascending (~31.7% to 40% of new deposits) |
Rather than rendering these traditional techniques obsolete, predictive computational modeling has integrated synergistically with experimental pipelines. In X-ray crystallography, accurate predictive AI models now serve as initial search models for molecular replacement, resolving the phase problem without the need for complex heavy-atom derivatization 6. In Cryo-EM workflows, rigid-body docking of AI-predicted domains into lower-resolution overall density maps greatly accelerates the atomic model building process for massive, heterogeneous macromolecular complexes 6.
Architectural Evolution of the AlphaFold Models
The introduction of the AlphaFold framework by Google DeepMind represented a fundamental paradigm shift in computational structural biology, transitioning the field away from physics-based energy minimization algorithms toward sophisticated deep learning neural network architectures.
AlphaFold 1 and the Shift to Deep Learning
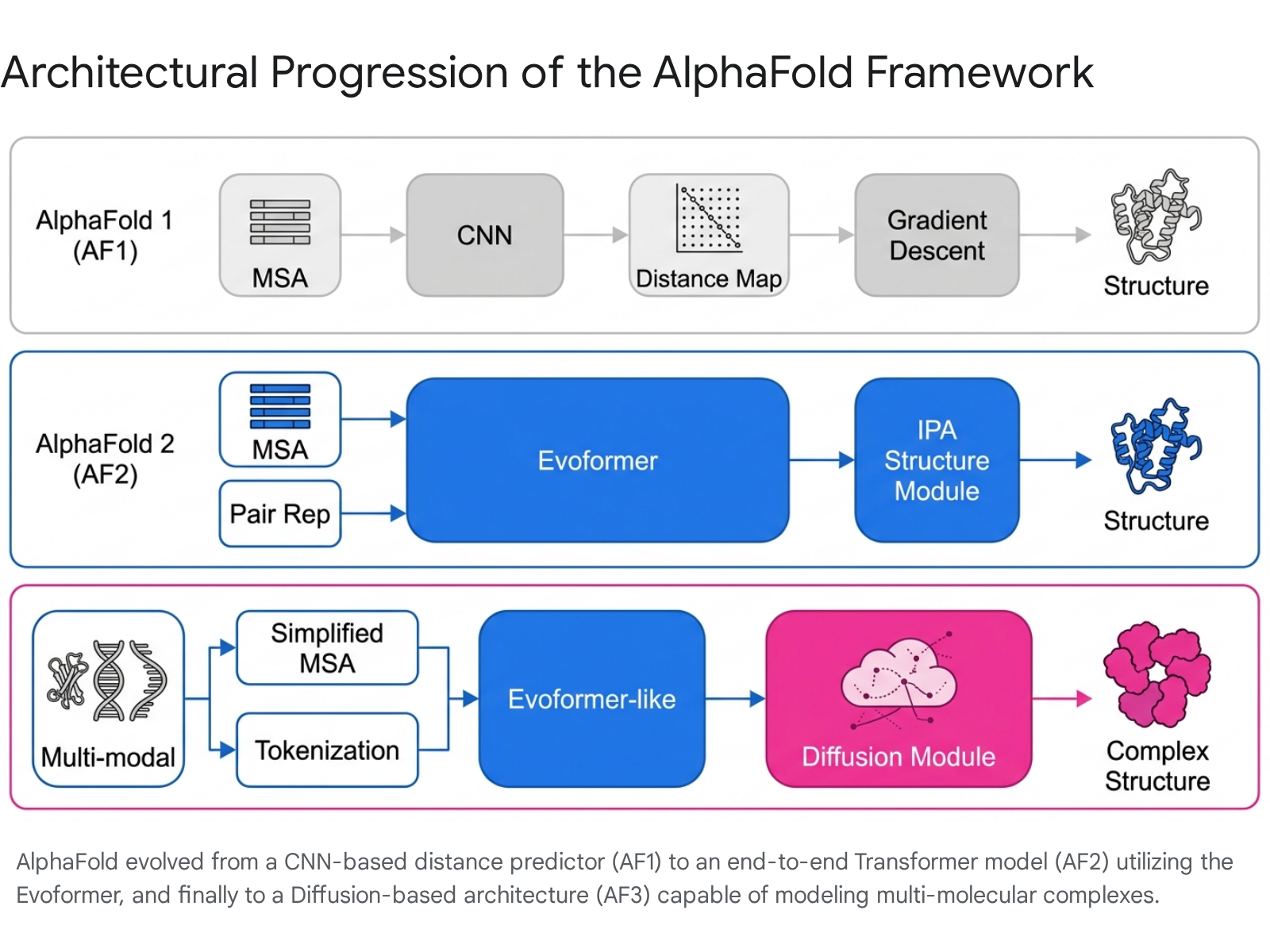
AlphaFold 1 (AF1) was introduced to the scientific community at the CASP13 blind experiment in 2018, representing a significant step forward by achieving a Global Distance Test (GDT) score of roughly 60% 1. The AF1 architecture utilized a deep convolutional neural network (CNN), a methodology traditionally optimized for two-dimensional image recognition tasks 110. The network was trained extensively on existing PDB structures to predict distance maps based on multiple sequence alignments. These distance maps functioned as probability distributions indicating the spatial proximity between pairs of amino acid residues 1.
Crucially, AF1 was not an end-to-end differentiable system that directly generated 3D coordinates. Instead, the predicted distance maps were utilized to mathematically construct a potential of mean force. This artificial energy landscape was subsequently optimized using a traditional gradient descent algorithm to generate the final three-dimensional topological structure 1. While AF1 outperformed competing methodologies, its reliance on CNNs and secondary optimization steps limited its absolute accuracy.
AlphaFold 2 and Transformer Architectures
AlphaFold 2 (AF2) represented a complete architectural redesign, introduced at CASP14 in 2020. Recognizing the limitations of CNNs, the DeepMind team abandoned convolutional approaches in favor of a Transformer-based architecture, relying heavily on attention mechanisms to learn which distinct parts of an input sequence were most evolutionarily and physically relevant 1. At CASP14, AF2 achieved a median GDT_TS score of 92.4, an unprecedented accuracy level widely considered to be statistically indistinguishable from experimental determination methodologies 11. The average atomic position error of AF2 predictions was measured at merely 1.6 Angstroms 11.
Unlike its predecessor, the AF2 architecture operates as an end-to-end neural network that outputs explicit atomic coordinates directly from primary amino acid sequences. Its core innovation is the Evoformer block. The Evoformer processes two primary inputs simultaneously: a one-dimensional multiple sequence alignment (MSA) representation and a two-dimensional pair representation (distance matrix) 1. Through repeated attention layers, these two representations continuously exchange information, updating each other throughout the learning process. This allows the network to synthesize evolutionary co-variation context against physical spatial constraints 1.
The refined representations are then passed to a dedicated Structure Module, which operates on a three-dimensional backbone using the pair representation and the target sequence. This module treats the protein as a "residue gas" - a mathematical space where N-C$\alpha$-C geometric triangles float freely as rigid bodies. These triangles are translated and rotated into a coherent, physically plausible backbone utilizing an "invariant point attention" mechanism 1. Finally, the resulting output is subjected to a recycling mechanism, wherein the predicted structural coordinates are fed back through the Evoformer multiple times to resolve local stereochemical clashes and refine the thermodynamic potential of the final output 1.
AlphaFold Database and Research Adoption Statistics
To maximize the utility of AF2, Google DeepMind partnered with the European Bioinformatics Institute (EMBL-EBI) to launch the AlphaFold Protein Structure Database (AFDB) in 2021 127. Initially launched with approximately 350,000 structures representing the human proteome, the database was rapidly expanded. By 2022, the AFDB housed over 214 million predicted structural entries, encompassing nearly all known cataloged proteins across the biological spectrum sequenced in the UniProt database 1178.
The adoption metrics for the AFDB are staggering within the context of biological sciences. As of late 2025, the database has been actively utilized by over 3 million researchers spanning more than 190 countries 111215. Over one million of these active users are located in low- and middle-income countries (LMICs) 815. The AlphaFold methodology and outputs have generated over 43,000 direct academic citations, while more than 200,000 distinct research papers have incorporated elements of the AF2 framework into their computational or experimental methodologies 1115.
An independent econometric analysis conducted by the Innovation Growth Lab detailed the systemic impact of AF2 on experimental productivity. The report found that academic laboratories utilizing AF2 experienced an increase of over 40% in their submissions of novel experimental protein structures to the PDB, indicating that AI modeling helps researchers explore structural areas that were previously too difficult to access experimentally 12159. Furthermore, research building upon AF2 predictions was found to be twice as likely to be cited in downstream clinical articles and demonstrated a significantly higher probability of being cited in pharmaceutical patents compared to baseline works in structural biology 15. The database interface itself continues to undergo refinements, with the 2025 UniProt alignment update introducing dedicated domain summaries, interactive 3D viewers for complex structural interpretation, and integrated sequence alignments 1011.
AlphaFold 3 and Multi-Molecular Complex Prediction
While AF2 effectively solved the isolated single-chain protein folding problem, cellular biology is fundamentally driven by complex multi-molecular assemblies. Proteins rarely execute their biological functions in isolation; they interact dynamically with nucleic acids (DNA and RNA), small molecule ligands, post-translational chemical modifications, ions, and other protein chains to form operational machines 1213. To address this reality, Google DeepMind and its commercial spin-off Isomorphic Labs released AlphaFold 3 (AF3) in May 2024 131415.
Diffusion-Based Architecture
AF3 builds upon the foundational Evoformer principles established in AF2 but introduces critical architectural modifications to handle heterogeneous molecular environments. It utilizes an extended, generalized tokenization scheme capable of processing a much broader taxonomy of chemical entities. This allows the network to simultaneously parse and evaluate amino acid chains, nucleotide sequences, and SMILES strings representing small molecule ligands 1416. Concurrently, the MSA processing step was structurally simplified to minimize computational overhead and enhance processing efficiency across diverse input types 1424.
The most profound architectural shift in AF3 is the complete replacement of the AF2 coordinate-generating Structure Module with a novel Diffusion Module 1014. Diffusion models - the same underlying class of generative algorithms driving high-fidelity AI image synthesis - operate by iteratively removing noise from a corrupted system. In the context of AF3, the model begins with a highly noisy, randomized cloud of atomic coordinates. Guided by the highly refined sequence and pair representations from the upstream network, the diffusion module progressively denoises the coordinates over multiple steps. The system continually refines the positions of the atoms until a thermodynamically plausible, physically valid multi-molecular complex emerges, allowing the model to position diverse molecular entities without relying on rigid body constraints 1424.
Benchmarking Protein-Ligand Interactions
The transition from single-chain prediction to complex prediction necessitated rigorous benchmarking against specialized physical docking software and competing AI models, most notably the RoseTTAFold All-Atom model developed by the Baker Laboratory at the University of Washington. Accurately predicting protein-ligand interactions represents a historic challenge in computational chemistry, requiring the determination of not only the macro-fold of the protein receptor but the highly sensitive, atomic-level conformational shifts of the binding pocket as it accommodates a small molecule.
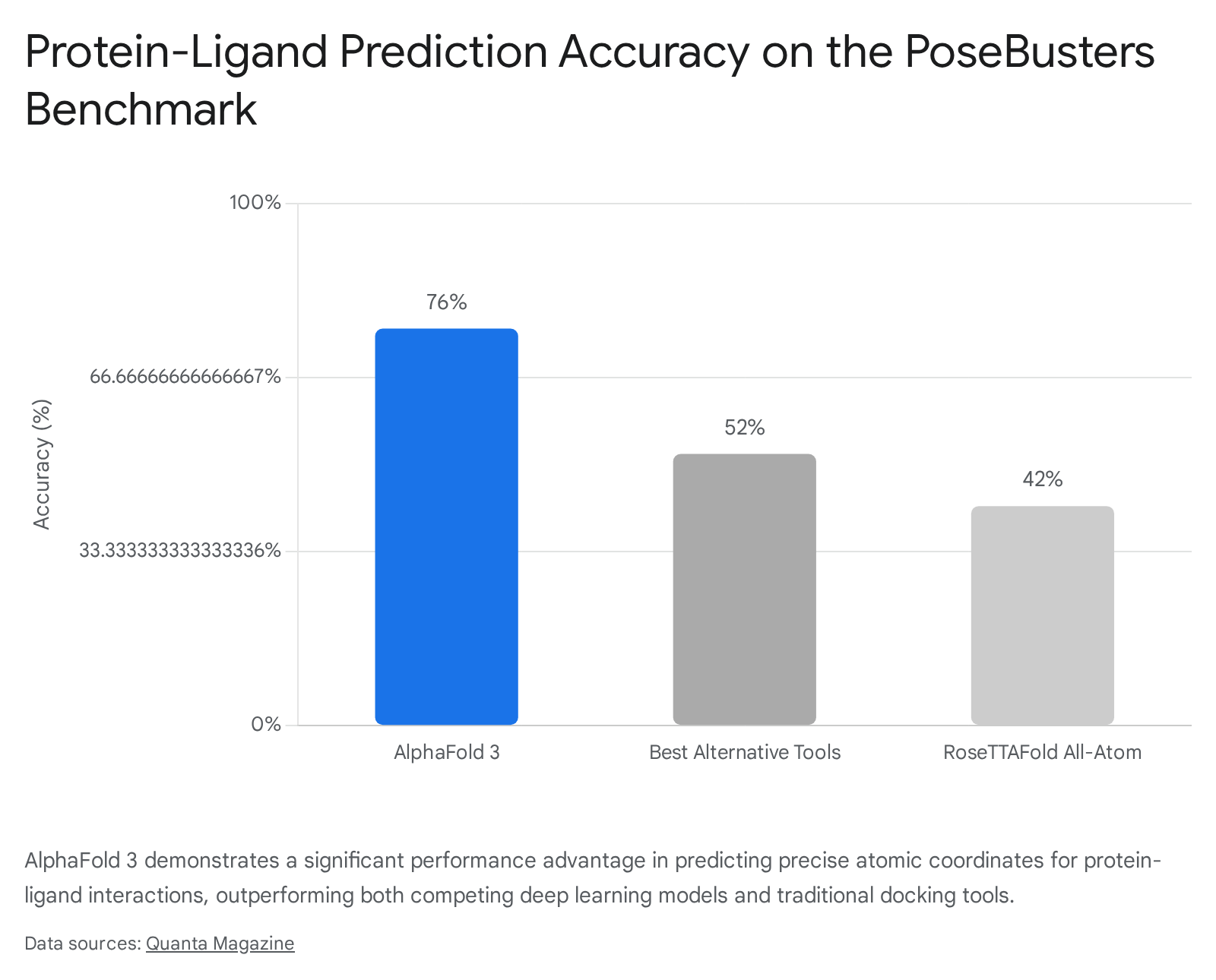
To evaluate this capability, researchers utilized the PoseBusters benchmark - a rigorous dataset of 428 diverse protein-ligand structures released after the training cut-off for the models. On this dataset, AF3 achieved a ligand Root Mean Square Deviation (RMSD) success rate of approximately 76% 131718. This performance significantly outpaced traditional physics-based docking software (such as state-of-the-art AutoDock Vina, which scored 52%) and competing AI tools, with RoseTTAFold All-Atom achieving roughly 42% accuracy on comparable tasks 1317.
Despite these high-profile successes, independent assessments, such as the FoldBench evaluation, have highlighted contextual limitations within AF3's ligand docking capabilities. AF3 excels exceptionally at predicting static protein-ligand interactions where the binding pocket undergoes minimal conformational change upon binding (RMSD < 0.5Å compared to the apo, or unbound, state) 18. However, the model's accuracy degrades precipitously when tasked with modeling highly flexible ligands, unsaturated or branched-chain fatty acids, or when predicting the docking of "unseen ligands" 1518. When evaluated on unseen ligands - defined as molecules displaying a Tanimoto similarity score of less than 0.5 to any molecule within the model's training set - AF3's success rate dropped to 64.3%, indicating a heavy reliance on historical training data rather than pure physical reasoning 18.
Nucleic Acids and Antibody-Antigen Complexes
Beyond small molecules, AF3 demonstrates substantial improvements in modeling macromolecular assemblies, particularly protein-nucleic acid complexes and antigen-antibody interactions. In direct statistical comparisons using standardized biological datasets, AF3 surpasses RoseTTAFoldNA in both global and local distance prediction accuracy for protein-DNA and protein-RNA multimers 1419.
The prediction of ab initio single-chain RNA structures, however, remains highly complex. Unlike proteins, RNA sequences often lack the deep evolutionary co-variation signals necessary to train highly accurate attention networks. For isolated RNA monomers, specialized secondary models like trRosettaRNA have occasionally demonstrated higher global topological accuracy in blind CASP assessments, though AF3 maintains a distinct advantage in resolving the local atomic accuracy of RNA interaction interfaces 14.
Similarly, AF3 resolves historic deficiencies in immunological modeling. Predicting how an antibody binds to its corresponding antigen requires modeling the complementarity determining regions (CDRs) - hypervariable loops that dictate immune specificity. Because CDR loops are intrinsically flexible and evolve rapidly, they lack clear evolutionary templates. AF3's diffusion architecture significantly outperforms older models like AlphaFold-Multimer v2.3 across all metrics in antigen-antibody interface prediction, showing an accuracy rate roughly 33.3% higher than its predecessor 171920.
| Complex Classification | AlphaFold 3 (AF3) Performance Profile | Performance of Competing Models / Traditional Methods |
|---|---|---|
| Protein-Ligand Docking | ~76% success on PoseBusters. High precision for rigid pockets, lower for unseen or highly flexible ligands. | RoseTTAFold All-Atom (~42%). Traditional physics docking software like Vina (~52%). |
| Protein-Protein (Multimer) | Minor global improvements over AF-Multimer; substantial local accuracy gains at the binding interface. | AF-Multimer v2.3 shows comparable global topological accuracy but weaker local contact prediction. |
| Antigen-Antibody | Significant outperformance (33.3% higher accuracy) across all metrics relative to earlier AF versions. | Historically challenging for all models due to hypervariable CDR loops. |
| Protein-Nucleic Acid | Surpasses RoseTTAFoldNA globally and locally for DNA/RNA complexes. | Deep learning trails human-assisted classical modeling for complex RNA ab initio folding. |
| RNA Monomers | High local accuracy, but global topology can be inconsistent. | trRosettaRNA maintains higher global topological accuracy for isolated RNA chains. |
Transformation of Pharmaceutical Drug Discovery
The capability to predict ligand-protein binding interfaces with atomic precision has catalyzed systemic strategic shifts within pharmaceutical development pipelines. By circumventing the years-long timeline traditionally required to isolate, purify, and crystallize target proteins using X-ray crystallography or Cryo-EM, computational diffusion models allow structural biologists to initiate virtual screening and structure-based drug design (SBDD) almost instantly upon identifying a target sequence 213022.
Economic Metrics and Pipeline Acceleration
The economic implications for the biopharmaceutical sector are profound. Research and development costs within the pharmaceutical industry have risen precipitously, reaching an average of $2.23 billion per late-stage asset in 2024, driven by increasing clinical trial complexity and highly saturated, competitive therapeutic markets 2324. Concurrently, the industry faces an impending patent cliff, with top-selling drugs losing patent protection and jeopardizing over $300 billion in sales between 2026 and 2030 25. This environment dictates an industry mandate to employ "fail-fast" R&D approaches 25.
Predictive AI models serve as the foundation for this fail-fast strategy. Highly accurate in silico druggability assessments allow pharmaceutical firms to evaluate target viability and abandon unviable projects before initiating costly in vitro synthesis 26. Industry estimates, including comprehensive analyses by the McKinsey Global Institute, project that generative AI and agentic modeling workflows in life sciences could ultimately create between $60 billion and $110 billion in annual economic value 36. According to late 2025 surveys conducted by Bloomberg Intelligence covering pharmaceutical executives responsible for pipeline budgets, the integration of advanced AI models like AlphaFold into end-to-end development is projected to trim total R&D outlays by an average of 16% 36. Furthermore, executives forecast that these tools will compress typical preclinical discovery timelines, shaving between 6 to 18 months off the development cycle for new therapeutics 36.
Target Identification and Virtual Screening
Prospective academic studies demonstrate that predictive models are now robust enough for direct operational use in virtual screening. In a rigorous study published in the journal Science, researchers at The Rockefeller University utilized AlphaFold models to screen billions of chemical compounds against specific disease targets 21. The study found that for the targets examined, the AI-predicted structures guided structure-based virtual drug screening just as reliably as high-resolution experimental crystal structures 21. The researchers concluded that utilizing AlphaFold in this manner could expedite initial discovery projects by up to several years in approximately one-third of cases 21. The technology is particularly valuable for accelerating research into membrane proteins - such as G-protein-coupled receptors (GPCRs) - which are notoriously difficult to crystallize but represent the targets of a vast majority of modern pharmaceuticals 30.
Clinical Progression and Isomorphic Labs
In applied commercial contexts, the descendants of the AlphaFold architecture are moving rapidly toward the clinic. Isomorphic Labs, a biotech spin-off of Google DeepMind, operates a proprietary drug design engine termed IsoDDE, which builds upon the foundational AF3 architecture to provide enhanced predictive accuracy for drug modalities including small molecules, peptides, and biologics 3727. Leveraging this platform, Isomorphic Labs has actively applied generative chemistry models to its oncology and immunology pipelines.
By modeling complex interaction dynamics - such as how therapeutic antibodies bind to specific cellular antigens, or how novel small molecules dock into cryptic binding pockets - the company has significantly accelerated the lead optimization phase 3027. Through major partnerships with pharmaceutical giants including Novartis, Eli Lilly, and Johnson & Johnson, Isomorphic Labs advanced its first natively AI-designed molecules toward first-in-human clinical trials slated for 2025 and 2026, marking a critical milestone in the transition from computational prediction to real-world medicine 3727.
Applications in Neglected Tropical Diseases
Beyond highly capitalized commercial oncology pipelines, predictive structural modeling facilitates rapid advances against neglected and emerging global diseases. The Drugs for Neglected Diseases Initiative (DNDi) actively utilizes the AlphaFold database to identify previously unknown target molecules for diseases that disproportionately affect developing nations 39. For example, researchers utilize structural data to isolate molecular vulnerabilities in the Trypanosoma cruzi parasite, the causal agent of Chagas disease in the Americas, as well as the parasites responsible for leishmaniasis 39.
Furthermore, predictive models vastly accelerate drug repurposing by rapidly mapping existing, FDA-approved compounds against newly modeled pathogen proteins. During the study of SARS-CoV-2, the AlphaFold platform was deployed to identify potential inhibitors for the viral non-structural protein 6 (NSP6), rapidly recommending candidates for biological testing without waiting for the empirical crystallization of the viral target 22. In the fight against antimicrobial resistance, researchers utilizing AlphaFold successfully mapped the structure of bacterial resistance proteins in approximately 30 minutes - a task that had proven technologically unfeasible for the preceding decade 39.
Agricultural Biotechnology and Pathogen Resistance
The implications of high-accuracy protein structure prediction extend fundamentally into agricultural biotechnology, offering novel pathways to ensure global food security. Plant proteins dictate essential physiological processes, including photosynthesis, nutrient absorption, and complex environmental stress responses 2829.
Crop Resilience and Abiotic Stress Tolerance
As climate change introduces severe abiotic stress vectors - including extreme heat, prolonged drought, and soil salinization - accelerating crop adaptation is a global priority. Computational modeling provides botanists with deep mechanistic insights into plant resilience 42. For example, AF3 has been utilized to model massive multi-protein complexes like small heat shock proteins (sHSPs) 28. These sHSPs act as molecular chaperones, preventing vital cellular proteins from misfolding and aggregating into toxic clumps during severe thermal stress 28. By identifying and visualizing the stabilization interfaces of specific proteins like HSP20, bioengineers have successfully overexpressed these genes, resulting in enhanced root growth and germination rates in staple crops like rice under high salinity and extreme heat conditions 28.
Similarly, researchers leverage AI-generated structures to map complex plant immune signaling cascades. Models have successfully detailed the precise physical interaction between the master regulator of plant immune signals, NPR1, and transcription factors like TGA3 in the model organism Arabidopsis thaliana 28. The predictive models revealed exactly how hydrophobic amino acid residues in NPR1 align perfectly onto the concave structure of TGA3, triggering the transcription of defense genes and systemic acquired resistance 28.
Cross-Kingdom Pathogen Interactions
Biotic stress caused by viral, fungal, bacterial, and oomycete pathogens destroys substantial fractions of global agricultural yields annually. Adapted plant pathogens deploy vast arrays of small secreted proteins (SSPs) and effector molecules that penetrate plant cells to hijack molecular machinery, suppress host immunity, or extract nutrients 30. Determining the function of these hundreds of uncharacterized SSPs represents a massive bottleneck in agricultural virology and pathology.
AlphaFold-Multimer and AF3 allow researchers to conduct high-throughput in silico screening of thousands of protein-protein interactions (PPIs) to predict cross-kingdom binding interfaces. In a landmark computational screening, researchers analyzed 1,879 SSPs from seven different tomato pathogens against defense-related plant hydrolases 30. Out of 11,274 computationally modeled protein pairs, the AI accurately identified 15 previously unannotated SSPs that physically obstruct the active sites of plant defensive enzymes 30.
Crucially, the modeling revealed profound examples of convergent evolution and molecular mimicry 3031. The AI predicted that pathogens spanning entirely different microbial kingdoms - including the fungus Cladosporium fulvum, the bacterium Xanthomonas perforans, and the devastating oomycete Phytophthora infestans (the cause of the Irish potato famine) - all evolved entirely unique, structurally distinct effectors that convergently bind to and inhibit the exact same active site on the plant pathogenesis-related subtilase P69B 30. This cross-kingdom structural analysis identifies universal vulnerability points within plant immune systems, guiding the engineering of broad-spectrum resistant crops.
Viral Threats in the Global South
In the Global South, where localized staple crops face distinct and devastating viral pressures, structural predictions are deployed against indigenous agricultural threats. Cassava, the fourth largest source of calories globally, suffers immense yield losses from two primary diseases: Cassava Mosaic Disease (CMD), caused by nine species of begomoviruses transmitted by whiteflies, and Cassava Brown Streak Disease (CBSD), caused by ipomoviruses 3233. Concurrently, the Maize streak virus (MSV), an indigenous African geminivirus, causes up to $480 million in losses annually amongst smallholder cereal farmers in sub-Saharan Africa 34.
By utilizing AlphaFold structure predictions alongside sequencing data, agricultural researchers map the hypervariable and highly stable regions of viral coat proteins and receptor-binding domains 3235. Identifying structurally conserved, indispensable viral motifs provides the necessary targeting parameters for advanced biotechnology interventions. This structural groundwork directly facilitates the use of CRISPR/Cas9 systems, Transcription Activator-Like Effector Nucleases (TALENs), and Zinc Finger Nucleases (ZFNs) to precisely edit host-plant genomes, breeding robust, targeted viral resistance into local African cultivars without relying on lengthy traditional breeding cycles 333436.
Democratization of Structural Biology in the Global South
Beyond the immediate acceleration of drug and crop development, perhaps the most significant systemic shift caused by the AlphaFold revolution is the democratization of structural biology access, particularly for researchers in low- and middle-income countries (LMICs).
Overcoming Traditional Infrastructure Barriers
Historically, structural biology research was fiercely confined to elite academic institutions in the Global North possessing massive capital resources. Establishing an X-ray crystallography core or outfitting a Cryo-EM laboratory requires multi-million dollar investments, uninterrupted power grids, and highly specialized maintenance ecosystems 373839. Consequently, Africa remains the only continent globally without a domestic synchrotron light source 3739.
This severe infrastructure deficit historically restricted African researchers from leading molecular investigations into the continent's most pressing endemic challenges. While diseases such as malaria, tuberculosis, human African trypanosomiasis (sleeping sickness), and Lassa fever exact their greatest toll in Africa, the molecular research required to combat them was predominantly exported to Western laboratories 374041.
The open-source deployment of the AlphaFold Protein Structure Database - and free access to non-commercial prediction servers like Google ColabFold - effectively bypasses these historic geographic and economic barriers 3941. As of 2025, over one million of the AFDB's active users are located in LMICs 815. By replacing capital-intensive hardware requirements with high-accuracy in silico predictive software, African scientists are empowered to transition from acting merely as consumers of secondary biological data to becoming primary producers of transformative structural discoveries, directly leading research pipelines tailored to local epidemiology 38.
The BioStruct-Africa Capacity Building Framework
However, access to advanced algorithms is insufficient in isolation; the technology is only effective if scientists possess the biophysical expertise required to interpret, manipulate, and experimentally validate the AI outputs 39. To bridge this critical training gap, grassroots frameworks have emerged across the continent. BioStruct-Africa, a non-governmental organization operating in Kenya, Ghana, and Sweden, established a comprehensive, scalable framework to systematically train the next generation of African structural biologists 3842.
Founded in 2017, BioStruct-Africa integrates modern computational prediction directly with downstream experimental validation 3842. The organization hosts extensive, fully funded capacity-building workshops across the continent, including major events at the University of Ghana (2019), Mali (2022), Cameroon (2024), and Nairobi, Kenya (2025) 3842. These workshops train local researchers to formulate structural hypotheses, execute AlphaFold multimer algorithms to screen local pathogen targets, and critically, validate these models through remote access to European synchrotron facilities like the Diamond Light Source in the UK 373942.
Crucially, the organization operates on a highly scalable "Train-the-Trainer" model 3856. Graduates of earlier workshops return as lead facilitators for subsequent cohorts under the guidance of senior mentors 3856. This approach not only ensures sustainable capacity building - with an organizational goal of training a cohort of 1,000 early-career scientists over the next decade - but fosters a self-sustaining ecosystem of scientific leadership that operates independently of continuous Western intervention, focusing intensely on neglected tropical diseases and localized agricultural blights 3856.
Epistemological Limitations and the Open Source Landscape
Despite achieving unprecedented predictive milestones that have fundamentally altered biological research, the AlphaFold framework and its integration into the broader scientific community are not without friction. Persistent debates center on the epistemological limits of deep learning in physics, as well as highly controversial corporate licensing strategies.
Memorization Versus Generalization
A persistent, foundational technical critique centers on whether deep learning structure predictors truly "learn" the underlying biophysical energy landscapes of protein folding, or if they operate primarily as highly sophisticated pattern-recognition engines that interpolate over memorized PDB data 4344.
Peer-reviewed analyses highlight distinct predictive vulnerabilities when AI models attempt to fold metamorphic proteins, often referred to as fold-switchers. These are highly specialized proteins that have evolved to adopt two entirely distinct, stable conformations depending on cellular conditions or ligand binding states 1543. Evaluators point out that predictive models like AF3 struggle to generate both thermodynamic states. Instead, the network overwhelmingly generates only the conformation that is most abundant within the evolutionary multiple sequence alignment data 151943. This suggests that the network does not generate a true, responsive thermodynamic free-energy surface capable of tracking conformational shifts, but rather relies on data density 43.
Furthermore, as noted in comprehensive FoldBench structural assessments, while AF3 accurately predicts the binding sites for common pharmaceutical ligands, its performance drops significantly when confronted with "unseen" chemical entities lacking structural homology to the PDB training set 18. The model is also prone to "hallucination" when predicting intrinsically disordered protein regions, occasionally forcing unstable loops into rigid secondary structures that do not exist in solution 45. These data points indicate that while excellent generalization exists for highly constrained sequence architectures, the model's highest accuracy outputs remain fundamentally dependent on the memorization of evolutionary patterns embedded within its training corpora 4344.
Licensing Controversies and Access Restrictions
The release mechanism of AlphaFold 3 initiated severe backlash regarding corporate stewardship of foundational scientific tools. Unlike AF2, which was released entirely open-source with its underlying weights, the initial publication of AF3 in the journal Nature in May 2024 lacked both the source code and the trained model parameters 60466247. DeepMind opted for this restriction to explicitly protect the commercial competitive advantage of its drug-discovery spin-off, Isomorphic Labs 6062.
Instead of open access, external researchers were restricted to a hosted web server capped at ten predictions per day, which expressly prohibited predictions involving novel organic molecules akin to chemical probes and unapproved drugs 47. The scientific community sharply criticized this decision, arguing it severely violated the fundamental principles of peer review and scientific reproducibility, as independent researchers could not conduct high-throughput bulk analyses or rigorously verify the paper's broad claims regarding drug design 6247.
In response to sustained academic pressure and the rapid emergence of competing models, DeepMind pivoted. In November 2024, the company released the AF3 model code and mathematical weights to a public GitHub repository 624865. However, the licensing terms remain highly restrictive. Access to the model parameters is strictly gatekept; it is provided exclusively to researchers actively affiliated with non-commercial organizations (universities, non-profits, government bodies) and is explicitly restricted to non-commercial use 6266. Any research conducted utilizing AF3 on behalf of commercial pharmaceutical entities remains strictly prohibited under the user agreement 66.
Proliferation of Alternative AI Models
This restrictive licensing environment inadvertently catalyzed a massive acceleration in the development of fully open-source alternative AI models by both academic institutions and competing tech firms. Recognizing the danger of a monopolized structural biology ecosystem, competitors rapidly engineered platforms that approach or equal AF3's benchmarks.
Models such as RoseTTAFold All-Atom (from the University of Washington), Chai-1 (by Chai Discovery), OpenFold3, and the Boltz-1/Boltz-2 models have rapidly proliferated 101860. Boltz-2, for example, expanded directly upon AF3's conceptual architecture but incorporated empirical molecular dynamics ensembles into its training data to better capture protein flexibility, while uniquely offering binding affinity predictions 18. The rapid rise of these open-weights models guarantees that multi-modal structure prediction will remain highly accessible to the broader scientific and commercial communities, preventing any single corporate entity from restricting downstream drug development 1860.
Conclusion
The transition from physical biophysics to deep learning-driven predictive modeling represents one of the most consequential paradigm shifts in the history of molecular biology. By effectively solving the single-chain protein folding problem and expanding rapidly into the accurate prediction of complex multi-molecular assemblies, the AlphaFold framework and its contemporaries have systematically compressed experimental discovery timelines from years to hours. Despite persistent epistemological challenges regarding the distinction between thermodynamic generalization and data memorization, and ongoing friction surrounding corporate licensing, the technology has irreversibly altered the scientific landscape. By democratizing access to atomic-level structural resolution for researchers across the globe - particularly in the Global South - artificial intelligence has transitioned structural biology from being the costly endpoint of localized research to the universal starting point for rational therapeutic and agricultural design.