Immunosenescence and Age-Related Immune System Inflammation
The conceptualization of immunosenescence has undergone a profound transformation within the global immunological community, a paradigm shift heavily supported by consensus in leading peer-reviewed journals such as Nature Immunology and Immunity. Historically framed as a linear, progressive deterioration of host defense mechanisms - a simple "weakening" of the immune system - the aging immune architecture is now understood as a highly dynamic, fundamentally dysregulated network. This biological remodeling is characterized by a bimodal paradox: a diminished capacity to mount precise, high-affinity adaptive responses to novel pathogens, occurring simultaneously with a chronic, low-grade hyper-activation that predisposes the host to tissue-destructive autoimmunity and systemic inflammation.
To achieve a comprehensive understanding of human aging, the physiological scope of immunosenescence must be broadened beyond the classical confines of the adaptive immune system, encompassing the profound alterations within the innate immune compartment. Specifically, macrophage dysfunction, natural killer (NK) cell phenotypic alterations, and the pervasive influence of the Senescence-Associated Secretory Phenotype (SASP) are now recognized as the primary drivers of "inflammaging," a sterile inflammatory state inextricably linked to late-life morbidity. Furthermore, recent epidemiological and anthropological studies demonstrate that baseline inflammaging profiles and cytomegalovirus (CMV)-driven immunosenescence are not universal biological inevitabilities. Rather, they vary drastically across geographical and socioeconomic divides, particularly when comparing populations in heavily industrialized nations to non-industrialized indigenous cohorts.
As the translational gap between highly successful murine models and human clinical applicability becomes increasingly apparent - highlighted by recent clinical hurdles and paradoxical failures in thymic regeneration strategies - the focus of clinical gerontology has shifted toward targeted, precision immune rejuvenation. Utilizing clinical trial and demographic data spanning from 2023 to 2026, this report exhaustively maps the physiological mechanisms of immunosenescence, the socio-geographical modulators of inflammaging, and the current landscape of clinical interventions aimed at resetting the biological immune clock.
The Bimodal Paradox: Immunodeficiency and the Amplification of Autoimmune Risk
The prevailing misconception that immunosenescence equates solely to immune frailty fails to account for the heightened incidence of autoimmune diseases, such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), universally observed in older populations 12. The aging immune system is not merely exhausting its functional reserves; it is actively remodeling in ways that systematically break down both central and peripheral tolerance mechanisms.
T-Cell Compartment Remodeling and the Naïve-to-Memory Inversion
The most well-documented and clinically impactful hallmark of adaptive immunosenescence is the drastic structural shift in the T-cell repertoire. Throughout early childhood and young adulthood, the thymus maintains a robust output of diverse naïve T cells, characterized by high concentrations of T-cell receptor excision circles (TRECs). Mathematical models of post-thymic proliferation and TREC dynamics indicate that this output and peripheral maintenance remain relatively stable up to the age of 20 3. However, progressive thymic involution - driven fundamentally by the chronological downregulation of the master transcription factor Forkhead box N1 (FOXN1) in thymic epithelial cells (TECs) - leads to a precipitous decline in the de novo generation of naïve T lymphocytes 456.
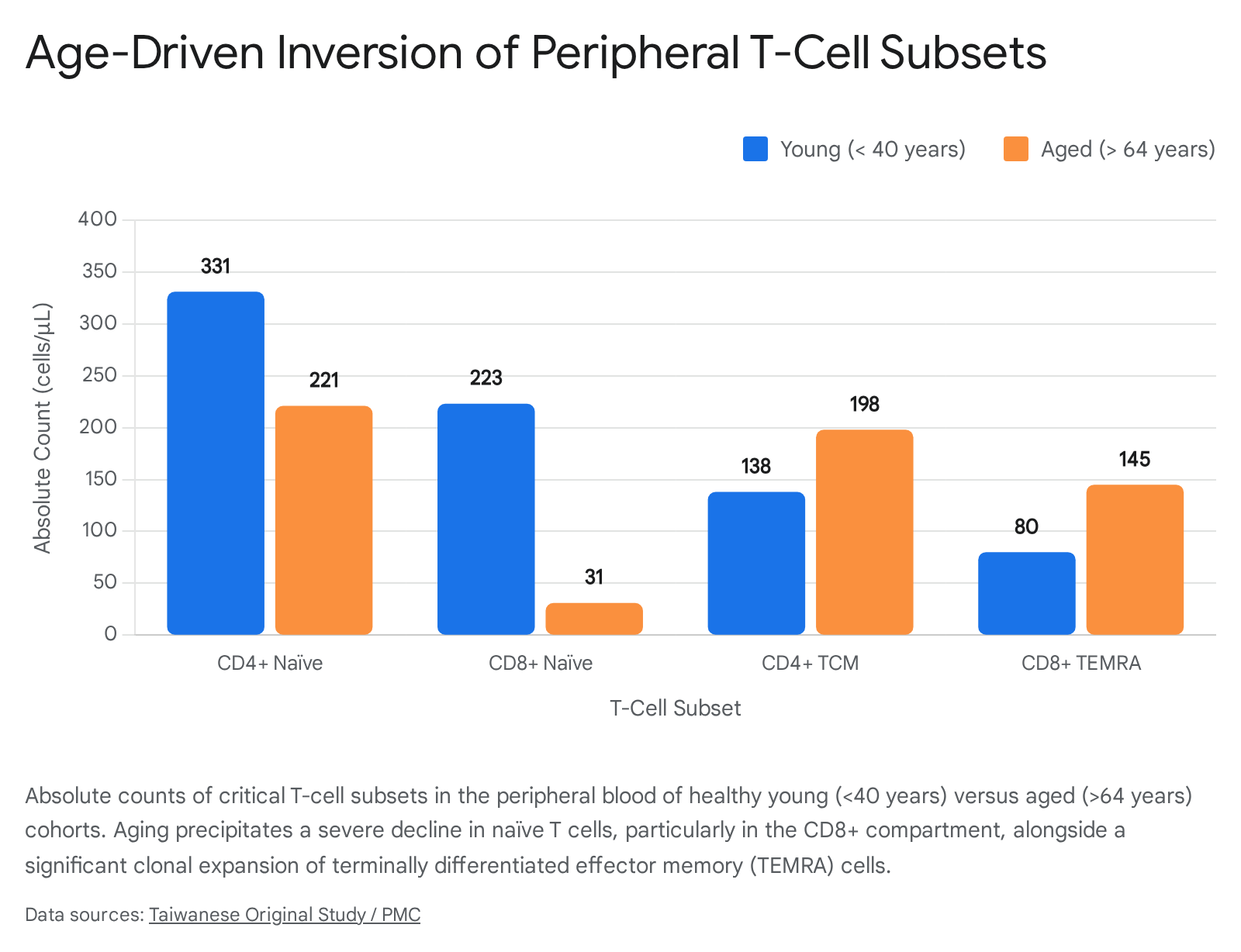
Quantitative analyses from recent meta-analyses and extensive cohorts in Taiwan and China provide stark boundaries for this decline. In the peripheral blood of healthy young individuals (under 40 years of age), absolute counts of CD4+ naïve T cells average 331 cells/μL, while CD8+ naïve T cells average 223 cells/μL 78. In older adults (over 64 years of age), this architecture collapses: CD4+ naïve counts drop to an average of 221 cells/μL, and CD8+ naïve T cell counts plummet to an average of just 31 cells/μL 8. To fill the immunological space left by reduced thymic output, homeostatic proliferation drives the massive, continuous clonal expansion of memory T cells 910.
This process is profoundly exacerbated by lifelong exposure to persistent viral infections, predominantly human cytomegalovirus (CMV). CMV continuously stimulates the immune system, leading to a phenomenon known as "memory inflation," wherein up to 20-30% of the entire CD8+ T-cell compartment may become dedicated to controlling this single latent virus, effectively crowding out the remaining naïve repertoire 1010. Consequently, the ratio of naïve to memory cells inverts. As CD8+ terminally differentiated effector memory (TEMRA) cells expand from an average of 80 cells/μL in youth to 145 cells/μL in old age, the CD4/CD8 ratio frequently drops below 1:1, establishing a widely recognized clinical immune risk phenotype 89.
The Collapse of B-Cell Repertoire Diversity and Clonal Dominance
A parallel and equally devastating deterioration occurs within the B-cell compartment. While total absolute B cell numbers experience only a modest decrease with advancing age - falling from an average of 244 cells/μL in young cohorts to roughly 217 cells/μL in the elderly - the qualitative functional decline is exponentially more severe 811. Baseline B-cell generation in the bone marrow is chronically impaired due to hematopoietic stem cell (HSC) lineage skewing, which progressively favors the production of myeloid progenitors over lymphoid progenitors 1213.
The most critical consequence of this age-related dynamic is the dramatic collapse in the diversity of the B-cell receptor (BCR) repertoire in late life. High-throughput next-generation sequencing of immunoglobulin heavy chain (IGH) transcripts reveals that while young individuals maintain a highly polyclonal repertoire characterized by a normal Gaussian distribution of complementarity-determining region 3 (CDR3) lengths, older individuals exhibit deeply restricted clonal diversity 141516. Aged repertoires are increasingly dominated by significant oligoclonal expansions, where a small handful of sequences give rise to massive, hyper-expanded B cell clones that monopolize immunological space 17.
Advanced diversity metrics rigorously quantify this collapse. For example, Shannon entropy analyses of BCR repertoires show significant drops in elderly patients - frequently falling from robust diversity scores above 0.640 in younger, healthy states to skewed distributions below 0.550 during aging or immune stress events 1920. Additionally, somatic hypermutation rates, while generally established early in childhood, fail to adequately diversify novel responses in the elderly due to the underlying accumulation of "Age-associated B cells" (ABCs) and a profound failure in class-switch recombination 1819. This loss of BCR entropy severely restricts the genetic substrate available for mounting novel, high-affinity antibody responses, providing a mechanistic explanation for the chronically impaired efficacy of seasonal influenza and pneumococcal vaccines in the elderly 1420.
The Autoimmune Trajectory: CD28- Effector Expansion and Treg Instability
The paradox of increased autoimmunity existing concurrently with systemic immune decline is largely driven by the accumulation of highly differentiated, senescent-like T cells that have shed the CD28 co-stimulatory molecule. These CD28- T cells, which accumulate prematurely and in massive numbers in conditions like rheumatoid arthritis and type 2 diabetes mellitus (T2DM), possess shortened telomeres, exhibit profound mitochondrial dysfunction, and display resistance to apoptosis 1. Most critically, they secrete extraordinarily high levels of pro-inflammatory cytokines such as Interferon-gamma (IFN-γ) and Tumor Necrosis Factor-alpha (TNF-α) 1. Because they bypass standard co-stimulatory checkpoint requirements for activation, these cells are easily triggered to attack host tissues, driving joint destruction in RA and exacerbating pancreatic β-cell dysfunction in T2DM 1.
Furthermore, regulatory T cells (Tregs) - the immune system's primary enforcers of peripheral tolerance - suffer severe age-related functional impairments. While the overall absolute number of Tregs often paradoxically increases with age despite thymic involution, their lineage stability is severely compromised 218. Epigenetic drifts and highly inflammatory microenvironmental shifts lead to the loss of FoxP3 expression in these populations, generating rogue "ex-FoxP3" cells 2. These formerly suppressive cells acquire pathogenic effector-memory phenotypes and begin actively secreting inflammatory cytokines like IL-2 and TNF-α, directly fueling autoimmune pathogenesis and irrevocably breaking the fragile immune homeostasis of the elderly host 218.
Expanding the Scope: Innate Immunity, Myeloid Skewing, and the SASP Axis
To comprehensively map the landscape of immunosenescence, pathophysiological analysis must transcend adaptive immunity to encompass the innate immune system. As the front line of defense and the primary coordinator of all downstream inflammatory responses, the innate immune system's dysregulation fundamentally shifts the aging body into a state of chronic, sterile inflammation.
Macrophage and NK Cell Dysfunction
The aforementioned age-related skewing of hematopoietic stem cells (HSCs) heavily favors the production of myeloid progenitor cells 1218. Yet, despite this numerical preservation or even relative increase in circulating myeloid cells, their functional integrity is deeply compromised. Aged macrophages exhibit markedly reduced phagocytic capacity, severely impairing the efficient clearance of apoptotic cells, misfolded proteins, and invading pathogens 1213. Furthermore, macrophage polarization shifts abnormally; aged macrophages struggle to transition smoothly between the pro-inflammatory (M1-like) state necessary for acute defense and the anti-inflammatory, pro-resolving (M2-like) state required for tissue repair. This regulatory failure results in a persistent, low-grade secretion of inflammatory mediators from the myeloid compartment, even in the complete absence of acute pathogenic infection 1213.
Similarly, Natural Killer (NK) cells - the innate compartment's crucial players for anti-viral defense and tumor surveillance - experience significant phenotypic and functional shifts throughout the lifespan. The aging process is marked by an expansion of the highly mature, terminally differentiated CD56dim cytotoxic subset, occurring alongside a perilous reduction in the immature CD56bright subset that typically regulates and coordinates broader immune responses 1213. Despite the numerical dominance of these CD56dim NK cells in the elderly, their actual per-cell cytotoxicity, signaling efficacy, and ability to proliferate in response to standard cytokine cues (such as IL-2 and IL-15) are markedly blunted, severely weakening overall immunosurveillance against emerging malignancies 1213.
The Senescence-Associated Secretory Phenotype (SASP) and Inflammaging
The functional decline of both innate and adaptive immune cells is inextricably linked to the broader biological phenomenon of cellular senescence. As immune and stromal cells accumulate DNA damage, telomere attrition, and metabolic stress, they enter a state of irreversible cell-cycle arrest. However, rather than undergoing silent, programmed apoptosis, these senescent cells develop a hyper-secretory, metabolically active profile known as the Senescence-Associated Secretory Phenotype (SASP) 121824.
The SASP secretome constitutes a highly toxic, complex cocktail of pro-inflammatory cytokines, chemokines, growth factors, and matrix-degrading metalloproteinases. Chief among these mediators are Interleukin-6 (IL-6) - frequently termed the "cytokine for gerontologists" due to its potent predictive value for all-cause age-related mortality - as well as TNF-α, Interleukin-1 beta (IL-1β), and C-reactive protein (CRP) 212223. In healthy young individuals, baseline serum levels of these cytokines are near zero, rapidly spiking only during acute infectious challenges. In the elderly, however, baseline levels are constitutively elevated. Clinical thresholds indicate that an IL-6 serum level exceeding 2 ng/L is considered high risk, a threshold frequently crossed in seemingly healthy elderly populations, manifesting the phenomenon termed "inflammaging" 182425.
This chronic, low-grade elevation has devastating, cascading systemic consequences. Elevated IL-6 and TNF-α act systemically to drive insulin resistance, promote endothelial dysfunction and lipid deposition in cardiovascular disease, and exacerbate profound neuroinflammation 12627. In the central nervous system, systemic inflammatory signals cross the increasingly permeable aging blood-brain barrier to activate senescent microglia and astrocytes, accelerating neuronal loss in Alzheimer's and Parkinson's diseases 27. Crucially, the SASP creates a self-perpetuating vicious cycle: the inflammatory cytokines secreted by a small focal point of senescent cells act via paracrine signaling to induce premature senescence in neighboring healthy cells, exponentially accelerating systemic tissue degeneration across the organism 1232.
Table 1: Comparative Analysis of Young vs. Aged Immune Profiles
| Immune Compartment | Specific Metric / Cell Type | Young Profile (<40 Years) | Aged Profile (>65 Years) | Underlying Mechanism & Clinical Impact |
|---|---|---|---|---|
| Adaptive (T Cells) | Naïve T Cell Output (Absolute) | High (CD4+: ~331 cells/μL; CD8+: ~223 cells/μL) | Severely Depleted (CD4+: ~221 cells/μL; CD8+: ~31 cells/μL) | Thymic involution; severely limits capacity to respond to novel pathogens and vaccines 48. |
| Adaptive (T Cells) | Memory & Effector T Cells | Regulated, functional homeostasis (CD8+ TEMRA: ~80 cells/μL) | Massive clonal expansion (CD8+ TEMRA: ~145 cells/μL) | CMV-driven memory inflation and CD28- accumulation; drives tissue destruction and autoimmune risk 189. |
| Adaptive (B Cells) | BCR Repertoire Diversity | High Shannon entropy (>0.640), diverse specificities | Collapsed diversity, heavy oligoclonal expansion | Impaired somatic hypermutation and class-switching; poor antibody affinity and vaccine efficacy 14151920. |
| Innate (Macrophages) | Phagocytosis & Polarization | Efficient clearance, balanced rapid M1/M2 transitions | Impaired phagocytosis, persistent pro-inflammatory bias | Reduced clearance of cellular debris; sustains chronic inflammation and tissue damage 1213. |
| Innate (NK Cells) | Subset Distribution | Balanced immature CD56bright and mature CD56dim subsets | Accumulation of mature CD56dim subset | Reduced per-cell cytotoxicity and proliferation potential; poor tumor immunosurveillance 1213. |
| Systemic Milieu | Baseline Cytokines (IL-6, TNF-α) | Low to undetectable in the absence of acute infection | Constitutively elevated (IL-6 frequently >2 ng/L) | SASP secretion drives multi-organ degeneration, neuroinflammation, and metabolic syndrome 182224. |
Geographic and Socioeconomic Modulators: The Myth of Universal Inflammaging
While immunosenescence and inflammaging have long been dogmatically considered universal hallmarks of human aging, recent cross-cultural and anthropological research spanning 2024 to 2025 has unequivocally upended this assumption. Comparative studies investigating populations in highly developed, industrialized nations versus those in developing, resource-limited, or indigenous settings reveal that immune aging trajectories are heavily dictated by the "exposome" - the comprehensive totality of environmental, lifestyle, and infectious exposures a population endures over a lifetime 2829.
The Industrialized Baseline vs. The Indigenous Exposome
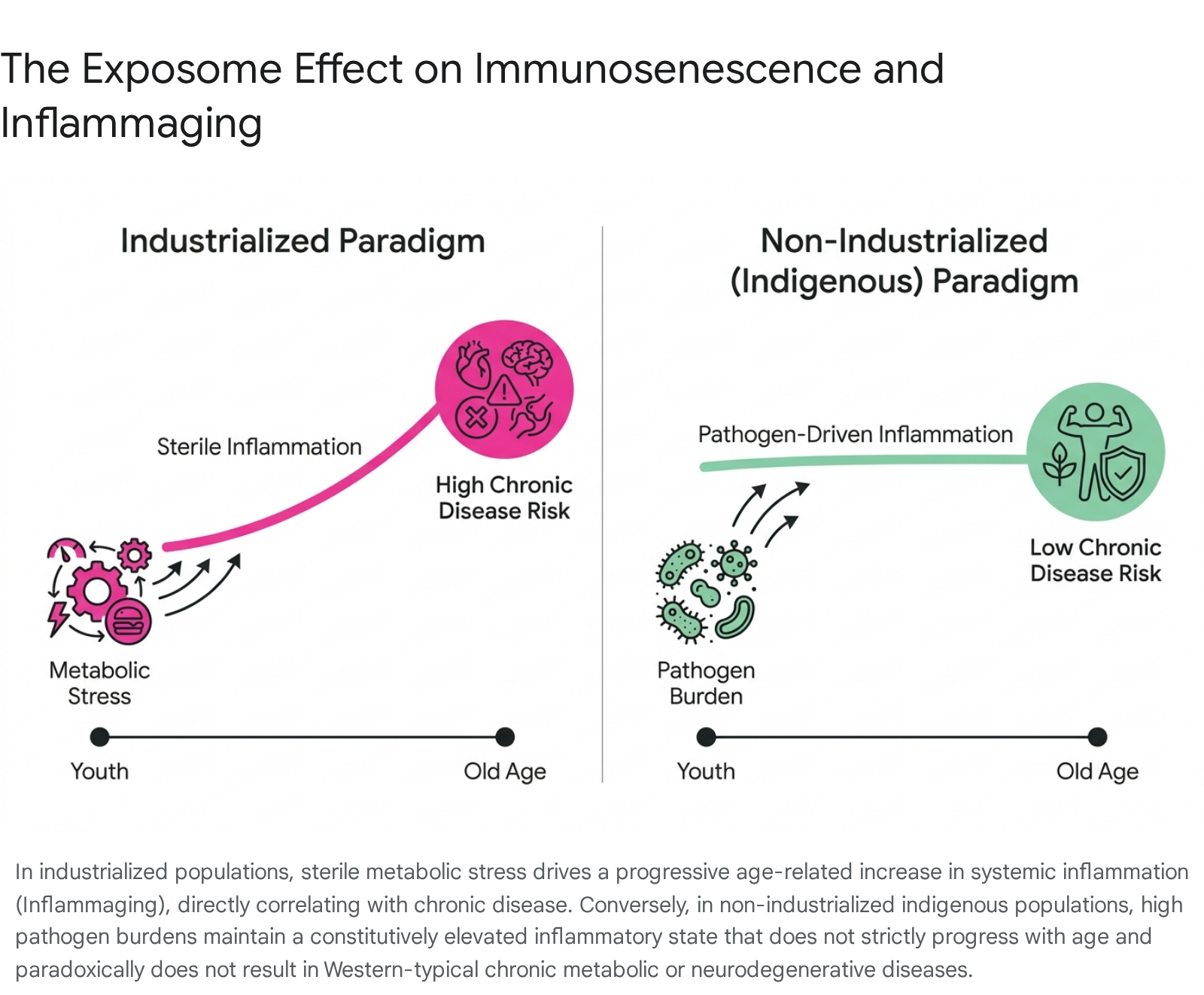
In industrialized settings (such as the United States, Western Europe, and Singapore), the recognized inflammaging profile is characterized by a gradual, age-progressive increase in sterile systemic inflammation. This age-correlated rise closely parallels the onset of chronic non-communicable diseases, including atherosclerosis, osteoarthritis, and metabolic syndrome 2829. Within these populations, inflammaging is largely a byproduct of industrialized lifestyles - driven by metabolic stress, visceral adiposity, and the unmitigated accumulation of senescent cells, rather than active pathogenic infection 129.
Conversely, exhaustive immunological investigations into indigenous, non-industrialized populations - most notably the Tsimane of the Bolivian Amazon and the Orang Asli of Peninsular Malaysia - demonstrate a radically divergent immunological paradigm. These populations exist in environments characterized by exceptionally high endemic pathogen burdens; for instance, approximately 66% of the Tsimane population and over 70% of the Orang Asli harbor at least one active intestinal parasitic or helminthic infection at any given time 28.
Consequently, young individuals in these indigenous cohorts exhibit baseline inflammatory biomarker levels (including IL-6 and CRP) that are constitutively elevated - mirroring or exceeding the highly elevated levels seen only in the elderly of industrialized nations 28. However, a critical physiological divergence occurs with advancing age: in the Tsimane and Orang Asli, these inflammatory markers do not increase progressively with chronological age 2829. Their systemic inflammation is entirely reflective of an ongoing, active infectious disease burden requiring constant immunological engagement, rather than an intrinsic, sterile, senescent aging process.
Uncoupling Inflammation from Chronic Disease and Socioeconomic Disparities
The most profound clinical revelation derived from these geographic comparisons is the uncoupling of inflammation from age-related pathology. In industrialized societies, high baseline inflammation in older adults guarantees a disproportionately high risk of chronic disease and frailty. Yet, in indigenous populations like the Tsimane, despite maintaining high circulating levels of inflammatory cytokines continuously throughout their lifespans, chronic age-related diseases such as heart disease, type 2 diabetes, and Alzheimer's are virtually absent 2829.
This data conclusively indicates that the specific source and context of immune activation dictate pathological outcomes. Inflammation driven by constant, natural immunological engagement with external pathogens (parasites, helminths) shapes the immune system in a fundamentally different manner than the sterile, metabolically-driven inflammation seen in Western societies 2930.
Furthermore, these dynamics manifest as health inequities within developed nations themselves. Studies investigating low-resource communities in the United States, such as the Mississippi Delta, reveal complex interactions where high rates of adiposity overlap with localized infectious exposures (such as Helicobacter pylori or intestinal parasites). In these disadvantaged cohorts, systemic inflammation (measured by CRP) correlates tightly with adiposity, while localized intestinal inflammation (measured by fecal calprotectin) relates to ongoing pathogen exposure, highlighting how socioeconomic status and local exposomes intricately compound immunosenescent risk factors, driving disparate health outcomes even within industrialized borders 30.
The Translational Gap: Clinical Hurdles in Thymic and Immune Regeneration
Recognizing the severe, cascading clinical consequences of immunosenescence, the biomedical field has aggressively pursued therapeutic strategies aimed at total immune rejuvenation. However, the path forward has been hampered by a significant translational gap between highly successful, elegant murine models and unpredictable, often disappointing human clinical outcomes.
The Anatomic Bottleneck: Thymic Involution and the FOXN1 Challenge
The thymus remains the primary anatomical bottleneck of the adaptive immune system. Restoring its function to re-establish naïve T-cell output and reset peripheral TCR diversity is the holy grail of immunogerontology.
In murine models, the forced upregulation of the transcription factor FOXN1 in aged thymic epithelial cells successfully reverses involution, rapidly restoring youthful thymic architecture and significantly boosting naïve T-cell export to the periphery 631. Recent state-of-the-art pre-clinical strategies have even utilized FOXN1-reprogrammed embryonic fibroblasts (FREFs) injected directly into the aged murine thymus, resulting in robust native thymus regrowth, reinforced negative selection, and a drastic reduction in autoreactive T-cell inflammation 31.
However, translating FOXN1-based therapies to human patients faces severe, multifaceted clinical hurdles. Anatomically, delivery mechanisms for gene therapy or cellular reprogramming directly into the heavily involuted, fibrotic, and fat-replaced human thymus are technologically daunting and highly invasive 32. Furthermore, extensive human genome sequencing, accelerated by newborn screening for Severe Combined Immunodeficiency (SCID), has revealed a vast array of human FOXN1 variants 33. While complete biallelic loss causes the well-documented nude/SCID phenotype (congenital athymia and alopecia), many monoallelic FOXN1 mutations act via dominant-negative mechanisms that unpredictably disrupt normal protein function 3334. This suggests that exogenous FOXN1 delivery in humans could perturb delicate TEC differentiation networks, potentially triggering severe lymphopenia or driving unconstrained, malignant epithelial proliferation (thymoma) rather than controlled regeneration 33.
The KGF (FGF7) Paradox
Keratinocyte Growth Factor (KGF, or FGF7) represents another profound example of the translational gap. KGF showed massive therapeutic promise in mice, consistently expanding TEC populations and protecting the delicate thymic microenvironment from severe radiation-induced damage 3536.
Yet, in recent high-profile human clinical trials involving patients with multiple sclerosis and HIV, recombinant human KGF administration failed to improve thymopoiesis. In one highly scrutinized trial following lymphodepletion, KGF treatment paradoxically reduced overall thymic output 35. Recent mechanistic studies from 2025 provide the explanation for this translational failure: while KGF treatment in humans rapidly induces the initial expansion of the epithelial compartment, it paradoxically downregulates FOXN1 and its downstream transcriptional targets (such as Dll4) in the critical early phases. This actively suppresses essential Wnt/β-catenin and Notch signaling pathways, arresting early thymocyte development at the β-selection checkpoint and resulting in an overall functional reduction of thymic mass and T-cell output 35. This failure distinctly highlights the extreme, poorly understood context-dependency of growth factor signaling within the human thymic microenvironment.
Clinical Interventions in Precision Immune Rejuvenation (2023 - 2026)
Where structural, anatomical thymic regeneration has stalled in human trials, pharmaceutical interventions aimed at systemic peripheral immune expansion and the targeted ablation of senescent cells have shown remarkable, practice-changing clinical success.
Systemic Rejuvenation and IL-7 Therapy
Interleukin-7 (IL-7) is a non-redundant, pleiotropic cytokine absolutely required for the survival, proliferation, and homeostasis of both naïve and central memory T cells 3738. Because native, endogenous IL-7 possesses a very short systemic half-life and occasionally triggers neutralizing anti-drug antibodies, recent clinical trials have utilized advanced, bioengineered formulations.
CYT107 (Efineptakin alfa), a fully glycosylated human recombinant IL-7 with an extended half-life, has dominated late-stage clinical research 3940. In human trials spanning 2024 to 2025, CYT107 has consistently demonstrated the ability to rapidly and durably restore CD4+ and CD8+ T cell counts in severely lymphopenic patient populations 4641. Crucially, unlike other immunomodulatory cytokines (e.g., IL-2), IL-7 achieves massive T-cell expansion without precipitating dangerous, highly lethal cytokine storms 42.
In a recent landmark prospective, double-blind, randomized trial published in JCI Insight (2025) involving critically ill COVID-19 patients suffering from severe virus-induced lymphopenia, CYT107 administration resulted in a highly significant 44% reduction in hospital-acquired secondary infections compared to placebo 42. This directly counteracts the core functional deficit of the immunosenescent phenotype by restoring a broad, functional T-cell repertoire capable of fighting opportunistic, nosocomial pathogens. These results have prompted ongoing, expansive Phase II/III trials in severe sepsis (the IRIS-7 trial) and broad oncology settings, where IL-7 is utilized to enhance the efficacy and persistence of CAR-T cell and immune checkpoint inhibitor therapies 374143.
Targeting the SASP: The Maturation of Senolytic Therapies
If IL-7 represents the accelerator for necessary immune cell proliferation, senolytics represent the targeted brakes applied to chronic inflammaging. Senolytics are novel pharmacological agents rationally designed to selectively induce apoptosis in senescent cells by transiently disrupting their Senescent Cell Anti-Apoptotic Pathways (SCAPs) 5051.
Dasatinib and Quercetin (D+Q): The combination of Dasatinib (a targeted tyrosine kinase inhibitor) and Quercetin (a plant flavonoid) remains the original and most extensively tested first-generation senolytic protocol. While highly effective at clearing senescent dermal fibroblasts and rejuvenating human skin grafts in complex human-mouse chimera models 44, systemic clinical outcomes have been deeply nuanced. A highly anticipated 2024 Phase II randomized controlled trial evaluated intermittent D+Q treatment in healthy postmenopausal women to combat osteoporosis. The therapy did not significantly reduce global bone resorption across the entire generalized cohort; however, precision genomic analysis revealed a major success: the therapy was highly effective at driving bone formation (measured via P1NP markers) only in the subset of patients who possessed a high baseline burden of senescent cells (quantified via T-cell p16 mRNA levels) 5153. This underscores a vital, emerging principle for all future senotherapeutics: clinical efficacy is highly dependent on pre-existing senescent burden, necessitating the rapid development of precise SASP biomarkers and "epigenetic immune clocks" for accurate patient stratification 153.
Fisetin and Next-Generation Localized Agents: Fisetin, a naturally occurring, highly potent flavonoid, has shown exceptional promise in preclinical models (e.g., progeroid mice) for clearing senescent cells and reducing SASP without the potential off-target chemotherapeutic toxicities associated with Dasatinib 3254. However, translating this to systemic human efficacy has proven difficult. A recent 2025 randomized Phase I/II clinical trial utilizing oral Fisetin to treat knee osteoarthritis found no significant benefit for pain, physical function, or cartilage health 55. Researchers attribute this failure primarily to the drug's inherent hydrophobicity and poor systemic oral bioavailability 55.
Consequently, the biotechnology sector is pivoting toward second-generation, highly targeted, localized senolytics. For instance, in 2024 and 2025, Unity Biotechnology reported highly positive topline results from their Phase 2b ASPIRE trial for UBX1325, a novel Bcl-xL inhibitor. By injecting the senolytic directly into the eye to clear senescent vascular cells, UBX1325 successfully improved visual acuity in patients with diabetic macular edema (DME), effectively achieving localized tissue rejuvenation while completely bypassing the risks and dilution of systemic administration 5657. Furthermore, next-generation research is currently exploring immune-based senolysis, engineering chimeric antigen receptor (CAR) T cells to specifically target senescence-surface markers like uPAR or GD3 ganglioside to allow the immune system to clear its own senescent cells 24.
Epigenetic Reprogramming and Immune Clocks
Alongside pharmacological interventions, systemic hormonal and metabolic reprogramming trials have advanced significantly. The highly anticipated TRIIM-X protocol (an extension of the original TRIIM trial) utilizes a combination of recombinant human growth hormone (rhGH), dehydroepiandrosterone (DHEA), and metformin to induce global thymic fat involution and broad immune rejuvenation 58.
The success of these holistic interventions is increasingly measured not just by cell counts, but by "epigenetic immune clocks" - advanced computational algorithms that quantify DNA methylation patterns across the genome to determine the true biological age of the immune system. These clocks, which correlate tightly with CD28- T cell accumulation and innate myeloid skewing, provide an incredibly precise biomarker for T2DM risk, autoimmune progression, and the actual biological efficacy of senolytic and rejuvenating therapies, bridging the gap between theoretical research and measurable clinical endpoints 145.
Table 2: Summary of Proposed Immune Rejuvenation Therapies and Clinical Status
| Therapeutic Agent | Mechanistic Target | Target Condition / Goal | Current Clinical Phase | Recent Findings (2023 - 2026) & Limitations |
|---|---|---|---|---|
| KGF / FGF7 | Thymic Epithelial Cell (TEC) expansion | Reversal of thymic involution | Phase II (Failed/Halted in human trials) | Fails in humans due to paradoxical early downregulation of FOXN1 and Notch signaling, arresting thymopoiesis 3536. |
| CYT107 (rhIL-7) | IL-7 Receptor; CD4/CD8 T-cell proliferation | Severe Lymphopenia, Sepsis, Cancer, COVID-19 | Phase II / Phase III | Highly effective and safe; demonstrated a 44% reduction in secondary hospital-acquired infections in critical viral patients 4142. |
| Dasatinib + Quercetin | SCAP Network (Broad Senolysis) | Systemic SASP reduction, Frailty, Osteoporosis | Phase II | Modest global efficacy; however, demonstrates highly significant physiological rejuvenation specifically in patient cohorts with a high baseline p16 senescent burden 514453. |
| Fisetin | p53/p21 pathway (Flavonoid Senolysis) | Systemic aging, Osteoarthritis, Inflammation | Phase II | Excellent safety profile, but recent OA trials show poor efficacy, largely attributed to extreme hydrophobicity and poor systemic oral absorption 3255. |
| UBX1325 (Unity) | Bcl-xL inhibitor (Localized Senolysis) | Diabetic Macular Edema (DME) | Phase IIb (ASPIRE Trial) | Achieved significant visual acuity gains via targeted, localized clearance of senescent vascular cells, effectively bypassing systemic toxicity 5657. |
| TRIIM-X Protocol | Epigenetic reprogramming (GH/DHEA/Metformin) | Global immune aging, Thymic fat involution | Phase II / Ongoing | Seeks to definitively prove systemic multi-pathway immune rejuvenation and validate reversal of biological age via advanced epigenetic immune clocks 158. |
Conclusion
The biological and clinical characterization of immunosenescence has evolved far beyond the simplistic, linear model of a depleting cellular reservoir. It is a highly complex, bimodal condition defined by the catastrophic loss of naïve antigen-responsive capacity juxtaposed violently against a chaotic, auto-reactive hyper-activation of memory lymphocytes and innate macrophages. The toxic accumulation of senescent immune cells and their associated highly inflammatory SASP drives the systemic "inflammaging" that underpins the vast majority of age-related morbidity - ranging from metabolic syndrome to neurodegeneration - in industrialized societies.
Crucially, cross-geographical and anthropological studies have definitively proven that the trajectory of inflammaging is not an immutable biological law, but is highly susceptible to modulation by the environmental exposome. This inherent biological plasticity provides the definitive foundation for pharmacological immune rejuvenation. While attempts at structural, anatomical thymic regeneration via growth factors (KGF) and genetic reprogramming (FOXN1) remain severely bottlenecked by complex translational hurdles and unpredictable human microenvironmental feedback loops, systemic functional interventions are advancing rapidly. Extended half-life cytokines like IL-7 (CYT107) are demonstrating profound clinical utility in rescuing patients from fatal lymphopenia and secondary infections. Simultaneously, the rapid maturation of senolytics - evolving from broad systemic D+Q cocktails to highly targeted, localized inhibitors like UBX1325 and CAR-T immune-senolysis - highlights the future of precision gerontology. Ultimately, arresting immune aging will require a sophisticated combinatorial approach: clearing the toxic burden of senescent cells to halt systemic inflammaging, while simultaneously providing the precise exogenous cytokine signals necessary to safely rebuild a diverse, naïve lymphocyte repertoire.