Extracellular matrix aging and organ dysfunction
The extracellular matrix (ECM) is a highly dynamic, three-dimensional macromolecular network that provides essential structural support, biomechanical resilience, and biochemical signaling cues to resident cells across all tissues. Historically conceptualized as an inert physical scaffold, the ECM is now recognized as an active and primary determinant of cellular identity, morphological homeostasis, and organ-level functionality 12. The complete proteome of the ECM, formally termed the matrisome, accounts for approximately 4% of the protein-encoding genome in mammals. This specialized subset of the proteome comprises structural core components such as collagens, elastins, glycoproteins, and proteoglycans, alongside an associated matrisome of remodeling enzymes, affiliated proteins, and secreted soluble factors 34.
During the biological aging process, the precise equilibrium required for matrix synthesis, enzymatic crosslinking, and proteolytic degradation is disrupted. This dysregulation subjects the ECM to progressive, often irreversible structural deterioration. The aging matrix is typically characterized by fibrillar collagen fragmentation, systemic elastin degradation, aberrant non-enzymatic protein crosslinking, and the aggressive accumulation of advanced glycation end-products (AGEs) 567. These profound biochemical and mechanical alterations shift the matrix from a supportive, homeostatic microenvironment to a primary driver of tissue dysfunction. An aged ECM actively fuels chronic local inflammation, initiates cellular senescence, and drives progressive fibrotic remodeling 89. The resulting age-dependent ECM signature - or "matreotype" - is now heavily implicated in the pathogenesis of numerous age-related syndromes, ranging from cardiovascular disease and pulmonary hypertension to neurodegeneration and metastatic cancer 310.
Biochemical Drivers of Matrix Aging
The systemic aging of the extracellular matrix is propagated by a confluence of spontaneous non-enzymatic reactions and dysregulated enzymatic processes. These modifications fundamentally alter the biomechanical properties of the tissue scaffold, impairing intrinsic elasticity, promoting aberrant mechanotransduction pathways, and restricting physiological matrix turnover.
Non-Enzymatic Glycation and Advanced Glycation End-Products
A principal molecular driver of ECM aging is the progressive accumulation of non-enzymatic crosslinks mediated by advanced glycation end-products. The formation of AGEs occurs via the Maillard reaction, a non-enzymatic condensation process wherein reducing sugars, such as glucose and fructose, react with the free amino groups of long-lived matrix proteins 1112. This initial nucleophilic addition yields a chemically unstable Schiff base. While Schiff bases form rapidly and are highly reversible, they subsequently undergo slow molecular rearrangements to form more stable Amadori products 11. Over weeks to months, these intermediates undergo further irreversible oxidation, dehydration, and degradation to form stable AGEs, prominently including pentosidine, glucosepane, and N-carboxymethyl-lysine (CML) 1112.
The accumulation of AGEs drastically alters the structural and biological properties of tissue proteins. Fibrillar collagens, which possess exceptionally slow physiological turnover rates, act as primary biochemical sinks for time-dependent glycation 12. AGEs form persistent divalent crosslinks between neighboring collagen fibril residues. This process increases macroscopic tissue brittleness and renders the collagen matrix highly resistant to normal proteolytic degradation by endogenous matrix metalloproteinases (MMPs) 1113.
Beyond physical stiffening, AGEs actively drive cellular pathology by interacting with the receptor for advanced glycation end-products (RAGE) expressed on the surface of resident fibroblasts, endothelial cells, and immune cells. The AGE-RAGE interaction initiates a cascade of deleterious intracellular signaling events, centrally involving the activation of nuclear factor-κB (NF-κB). NF-κB translocation prompts the transcription and release of pro-inflammatory cytokines, chemokines, and reactive oxygen species (ROS), establishing a vicious cycle of oxidative stress that exacerbates continuous matrix damage 1516.
Dysregulation of Enzymatic Crosslinking and Lysyl Oxidase
While non-enzymatic glycation represents a pathological form of crosslinking, enzymatic crosslinking is strictly essential for normal tissue development, matrix assembly, and macroscopic tensile strength. The lysyl oxidase (LOX) family of copper-dependent amine oxidases - which includes LOX and the lysyl oxidase-like enzymes LOXL1 through LOXL4 - catalyzes the oxidative deamination of specific lysine and hydroxylysine residues situated in the telopeptide regions of fibrillar procollagens and tropoelastin 714. This highly regulated enzymatic process converts these residues into reactive aldehydes, specifically allysine and hydroxyallysine. These intermediates subsequently undergo spontaneous condensation to form immature divalent crosslinks, eventually maturing into stable trivalent crosslinks that stabilize the matrix 1415.
In youth and during functional tissue repair, LOX-mediated crosslinking provides necessary biomechanical stability. However, normal aging introduces a profound dysregulation in LOX activity and spatial distribution. Crucially, the progression of non-enzymatic glycation directly interferes with enzymatic crosslinking. Recent proteomic mapping indicates that the site-specific glycation of helical domain lysine residues competitively inhibits the normal LOX-mediated crosslinking pathway. For example, in type I collagen, non-enzymatic glycation selectively targets the exact helical domain lysine sites - such as α1(I)K87, α1(I)K930, α2(I)K87, and α2(I)K933 - that are strictly required for normal lysyl oxidase-controlled crosslinking 12.
This direct competitive overlap dictates that age-related AGE accumulation actively hinders functional, repair-oriented crosslink formation. The result is a mechanically paradoxical tissue state: tissues become biochemically stiffened and brittle due to AGEs, yet they remain mechanically weaker and highly prone to micro-tearing under load due to a lack of coordinated enzymatic repair 712.
| Feature | Enzymatic Crosslinking | Non-Enzymatic Crosslinking |
|---|---|---|
| Primary Mediators | Lysyl oxidase (LOX) and LOXL isoforms | Reducing sugars (e.g., glucose, fructose) reacting with free amine groups |
| Reaction Mechanism | Oxidative deamination of lysine/hydroxylysine into aldehydes | Maillard reaction forming Schiff bases, Amadori products, and eventually AGEs |
| Primary Target Residues | Telopeptide lysine and hydroxylysine residues | Helical domain lysine and arginine residues |
| Typical Structural Outputs | Allysine, hydroxyallysine, immature divalent and mature trivalent crosslinks | Pentosidine, glucosepane, N-carboxymethyl-lysine (CML) |
| Physiological Role | Essential for embryogenesis, tissue development, and homeostatic tensile strength | Pathological accumulation strictly associated with aging, diabetes, and oxidative stress |
| Biological Reversibility | Subject to normal turnover via matrix metalloproteinases (MMPs) | Structurally irreversible; confers high resistance to standard proteolytic degradation |
| Consequences in Aging | Frequently impaired by competing glycation, contributing to osteopenia and mechanical weakness | Increases macroscopic tissue brittleness, limits dynamic remodeling, triggers RAGE-mediated inflammation |
Table 1: Comparison of the biochemical mechanisms, targets, and physiological impacts of enzymatic versus non-enzymatic extracellular matrix crosslinking processes. 7111212131416
Matrix Metalloproteinase Imbalance and Elastin Degradation
Concurrently with aberrant crosslinking, the aging ECM suffers from dysregulated enzymatic fragmentation. The delicate balance between matrix deposition and clearance is maintained by matrix metalloproteinases and their endogenous inhibitors (TIMPs). During aging, specific collagenases (MMP-1, MMP-8, MMP-13) and gelatinases (MMP-2, MMP-9) exhibit altered spatial and temporal expression profiles, leading to uncoordinated matrix turnover 217. Aging also severely limits the bioavailability of endothelial nitric oxide (NO). This systemic NO depletion promotes the export of transglutaminase 2 (TG2) to the extracellular space, where it non-specifically crosslinks fibronectin and laminin complexes while concurrently activating latent transforming growth factor-beta (TGF-β) signaling, which drives further pathological collagen deposition 18.
Elastin, a highly stable, hydrophobic polymer responsible for tissue recoil in the vasculature, lungs, and skin, undergoes severe degradation with age. Unlike collagen, elastin is rarely synthesized in healthy adult tissues; thus, age-associated degradation by elevated elastases and specific MMPs is functionally permanent 1819. The resulting elastin fragments, referred to as elastokines, possess potent pro-inflammatory and chemotactic properties. Elastokines actively recruit circulating monocytes and lymphocytes to the tissue stroma, perpetuating a state of chronic, low-grade sterile inflammation commonly referred to as "inflammaging" 819.
Cellular Mechanotransduction and Matrix Stiffening
The macroscopic biomechanical stiffening of the aged extracellular matrix exerts profound phenotypic effects on resident cells. Cells continuously probe the physical properties of their local microenvironment through focal adhesions and transmembrane receptors, translating external mechanical forces into internal biochemical signals - a fundamental biological process known as mechanotransduction 2024.
Integrin Signaling and Cytoskeletal Tension
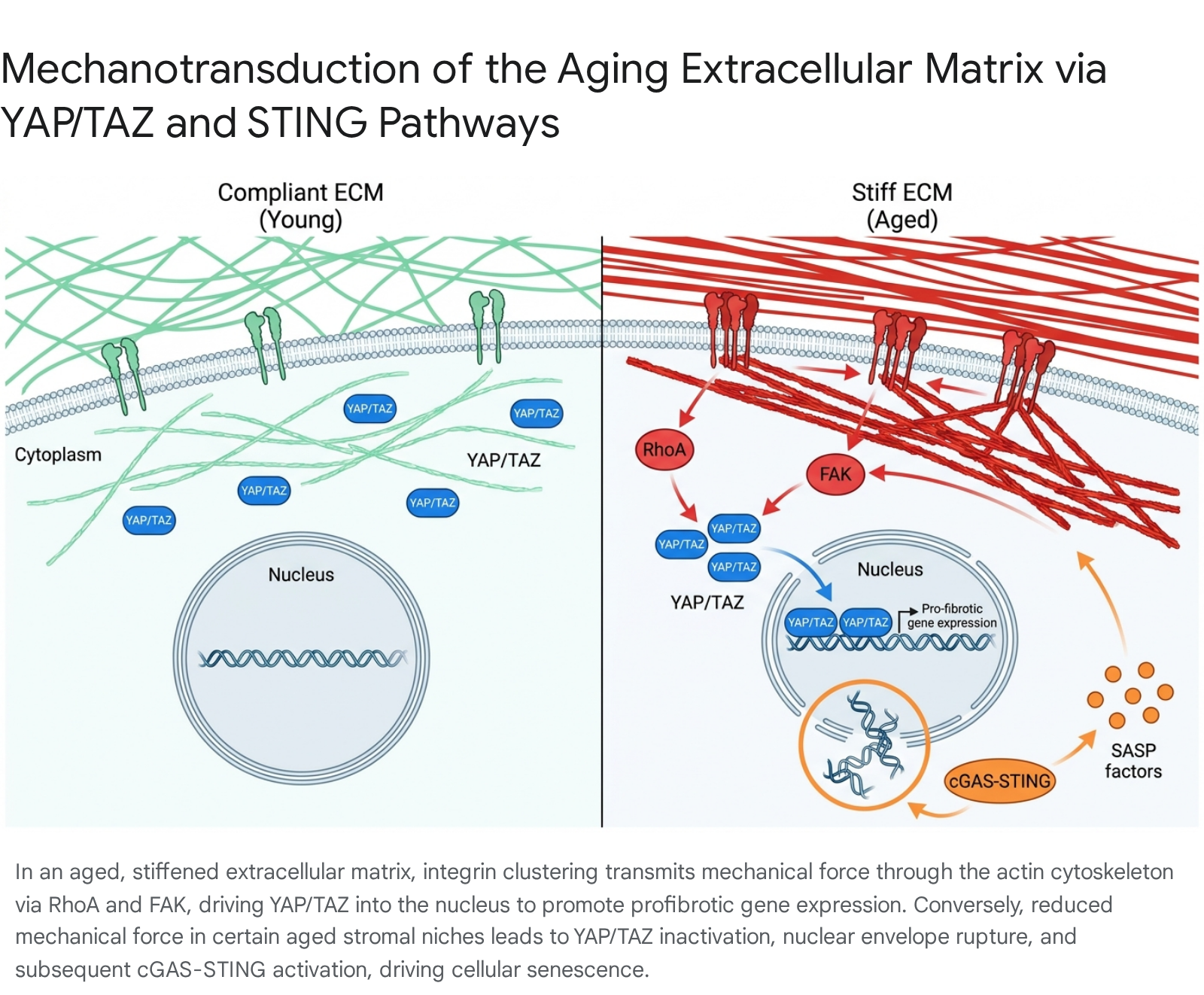
Integrins serve as the primary bidirectional mechanosensors physically bridging the extracellular matrix to the intracellular actin cytoskeleton. Upon encountering a stiffened, crosslinked matrix, integrin clustering at the cell membrane intensifies. This clustering triggers the rapid autophosphorylation of focal adhesion kinase (FAK) and the subsequent activation of small Rho GTPases, particularly RhoA and Rac1, leading to profound actin polymerization and increased intracellular cytoskeletal tension 2122.
The delicate balance between RhoA and Rac1 is crucial for maintaining cellular homeostasis. Genetic models exhibiting specific point mutations in mechanosensitive proteins, such as Filamin-A, demonstrate a disruption of this balance in favor of RhoA, altering the downstream trafficking of β1 integrins. This mechanism perfectly mirrors the aging phenotype, where elevated RhoA activity promotes pathological tissue stiffening, early-onset fibrosis, and hypercontractility 22. The mechanical tension generated by the actin cytoskeleton is transmitted directly across the nuclear envelope, where it governs epigenetic remodeling and transcriptional regulation 2123.
The YAP/TAZ Transcriptional Axis
Yes-associated protein 1 (YAP) and its paralog, transcriptional coactivator with PDZ-binding motif (TAZ), are the core downstream effectors of mechanotransduction and the Hippo signaling pathway. In young, highly compliant tissues, YAP and TAZ are persistently phosphorylated by LATS1/2 kinases, rendering them inactive and sequestered within the cytoplasm 521. However, in the context of a stiffened, aged, or fibrotic ECM, high mechanical cytoskeletal tension completely overrides this biochemical suppression. YAP and TAZ are rapidly dephosphorylated and translocate into the nucleus, where they complex with TEAD transcription factors to initiate broad epigenetic and phenotypic shifts 521.
Nuclear YAP/TAZ activation orchestrates a potent transcriptional program that heavily promotes cellular proliferation, aggressive ECM deposition (particularly collagens), and the upregulated expression of crosslinking enzymes like LOX 2024. This establishes a maladaptive, feed-forward loop: initial ECM stiffening activates YAP/TAZ, which in turn drives further profibrotic matrix deposition and enzymatic crosslinking, perpetually exacerbating the microenvironmental stiffness 2025.
This YAP/TAZ mechanosensing axis is heavily implicated as a core driver in oncogenic transitions. Ras-mediated oncogenic transformation typically requires a pre-existing stiff or fibrotic microenvironment to proceed. Cancer-associated fibroblasts heavily utilize YAP/TAZ signaling to increase intracellular isometric tension and continuously remodel the local ECM. This activity creates a dense, rigid tumor microenvironment that supports cancer cell survival, facilitates metastatic invasion, and creates a physical barrier against immune surveillance and chemotherapeutic penetration 82125.
Fibroblast Senescence and the Senescence-Associated Secretory Phenotype
The interplay between ECM aging and cellular senescence operates in a highly bidirectional manner. Cellular senescence - a state of stable, irreversible cell cycle arrest induced by accumulating molecular damage - is a primary, widely recognized hallmark of aging. Senescent cells progressively accumulate in aged tissues, particularly the skin, lung, and liver, where they secrete a highly complex cocktail of pro-inflammatory cytokines, chemokines, and matrix-degrading proteases known as the senescence-associated secretory phenotype (SASP) 58.
The SASP reinforces uncoordinated matrix degradation and sustains local chronic inflammation, drastically altering the structural microenvironment to induce senescence in neighboring, previously healthy cells through paracrine signaling. Conversely, the biomechanical and biochemical state of the ECM itself dictates entry into senescence. For instance, the age-related loss of α5-integrin and organized fibronectin in the dermal ECM abruptly disrupts the supportive niche environment necessary for standard fibroblast vitality. This specific extracellular loss directly promotes a senescence-like phenotype, characterized by a near-complete loss of cellular contractility and severe alterations in cytoskeletal organization 262728.
Recent mechanistic evidence uncovers an alternative, purely mechanobiological route to cellular senescence involving the cyclic GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING) pathway. In certain aged stromal cell populations, severe extracellular matrix disorganization results in profoundly reduced tissue mechanical force, leading to a precipitous loss of nuclear YAP/TAZ. This mechanical uncoupling compromises the physical integrity of the nuclear envelope, exposing genomic DNA directly to the cytoplasm. This misplaced cytoplasmic DNA is rapidly detected by cGAS, initiating robust STING activation, which independently drives the full SASP and promotes progressive tissue degeneration without the need for traditional telomere attrition or genomic DNA damage 5.
Endothelial Dysfunction and Vascular Rarefaction
In the vascular compartment, extracellular matrix aging exerts a devastating impact on endothelial cell physiology. Normal aging permanently disrupts the delicate endothelial balance between local vasodilators (such as NO) and potent vasoconstrictors, leading to chronically elevated oxidative stress and systemic mitochondrial dysfunction. This endothelial dysfunction exacerbates maladaptive ECM remodeling via continuous collagen deposition and uncontrolled elastin degradation 19.
Extensive mapping of vascular networks in aging models reveals marked microvascular attrition, or rarefaction, particularly localized in critical organ beds including the heart, kidney, and spleen 18. Microvascular endothelial cells extracted from aged subjects exhibit severely impaired angiogenesis when exposed to vascular endothelial growth factor (VEGF) in vitro. This progressive loss of microvascular density inherently decreases localized tissue perfusion, creating transient hypoxic zones that trigger further fibrotic matrix deposition, initiating an inescapable cycle of arterial stiffening and cardiovascular decline 18.
Organ-Specific Pathologies of Matrix Remodeling
While the fundamental molecular mechanisms of ECM aging are highly conserved across all bodily tissues, their ultimate pathological manifestations are heavily influenced by the distinct biomechanical demands and baseline structural matreotypes of specific organs.
Cardiovascular Stiffness and Myocardial Fibrosis
The cardiac extracellular matrix is a highly specialized, tissue-specific scaffold coordinating cardiomyocyte electrical coupling, gap junction integrity (via Cx43), and overall chamber mechanics 229. Resident cardiac fibroblasts act as the primary regulators of the myocardial ECM, continuously producing the collagen, fibronectin, and hyaluronan necessary for dynamic structural integrity 1729.
With advancing age, the heart predictably hypertrophies and becomes physically stiffer, a transition that severely impairs diastolic relaxation (compliance) and represents the primary physiological contributor to heart failure with preserved ejection fraction (HFpEF). This macroscopic stiffening is underpinned by reactive interstitial fibrosis, histologically characterized by an absolute increase in collagen I and III deposition, elevated fibronectin and vitronectin expression, and a concurrent, dangerous reduction in elastin and laminin β2 234. Furthermore, oxidative stress generated by age-associated mitochondrial dysfunction upregulates hypoxia-inducible factors (HIFs), which act to directly stimulate the transcription of profibrotic ECM genes across the myocardium 34.
A recently identified and underappreciated aspect of cardiac ECM aging involves the dynamic regulation of matrix fiber tension. Fibronectin fibers, which normally act as highly tensed mechanosensors physically bridging resident cells to the collagen network, drastically lose their mechanical tension in pathological aging states 17. In inflamed or dilated cardiomyopathies, these fibronectin fibers relax - a phenomenon likely driven by localized proteolytic cleavage by elevated MMP-2 and MMP-9 or the apoptotic loss of the anchoring fibroblasts. This relaxation of fibronectin tension compromises overall tissue mechanostasis and correlates strongly with systemic immune cell infiltration, elevated blood inflammatory markers, and the rapid deterioration of global cardiac output 17.
Pulmonary Hypertension and Microvascular Alterations
The pulmonary vasculature is exquisitely susceptible to mechanical alterations within its matrix. Remodeling of the pulmonary ECM through excessive collagen accumulation and simultaneous elastin degradation pathologically increases vascular tone and restricts necessary lung compliance 1820.
Clinical pulmonary hypertension (PH) is fundamentally driven by severe ECM stiffening localized to the small pulmonary arteries. The hyperproliferation of vascular smooth muscle cells (VSMCs) and activated myofibroblasts deposits a highly rigid matrix, which subsequently triggers the YAP/TAZ signaling cascade 20. The integrin-Gα13-RhoA axis senses the disturbed, highly pulsatile blood flow and extreme matrix rigidity, promoting rapid YAP nuclear translocation. Activated YAP/TAZ directly regulates the expression of glutaminase, fundamentally altering cellular metabolism to favor glutaminolysis and anaplerosis. This metabolic reprogramming provides the immense bioenergetic fuel necessary for the rapid, unchecked proliferation and migration of pulmonary artery endothelial and smooth muscle cells. Promising preclinical models demonstrate that targeted inhibitors against matrix crosslinking, such as specific LOX inhibitors, can successfully attenuate nuclear YAP/TAZ accumulation and heavily ameliorate terminal PH symptoms 20.
Dermal Atrophy and Ocular Remodeling
Dermal aging provides one of the clearest visual, histological, and functional readouts of progressive ECM decay. The structural integrity of the skin relies heavily on a dense, interlinked network of collagen and a highly organized fibronectin scaffold. Quantitative studies comparing young and aged dermis demonstrate a nearly 50% absolute reduction in fibronectin density in aged mammalian models 27.
Human dermal fibroblasts isolated from elderly human donors synthesize a profoundly sparser, highly disorganized fibronectin network in vitro when compared to fibroblasts from young subjects. This functional deficit is not merely a consequence of reduced overall cellular proliferation; it is mechanistically tied to an age-associated disruption in integrin trafficking. Specifically, aged fibroblasts exhibit a marked, irreversible reduction in α5-integrin surface expression. Because α5-integrin is strictly essential for binding and correctly assembling extracellular fibronectin, its persistent depletion disrupts the localized mechanosensory environment. This physical uncoupling precipitates a senescence-like transcriptional program in dermal fibroblasts, directly resulting in the chronic wound susceptibility, macroscopic tissue fragility, and impaired barrier function characteristic of aged skin 2627.
In the ocular space, specific ECM layers such as Dua's layer and the Descemet's membrane undergo similar structural aging. The decline in limbal stem cell (LSC) functionality and the disorganization of the underlying extracellular matrix severely compromise corneal epithelial homeostasis, necessitating advanced cell-sheet engineering therapies for ocular surface reconstruction 30.
Systemic Proteomics and Matreotype Aging Clocks
Advancements in highly multiplexed tandem mass spectrometry and targeted bioinformatics have recently enabled the precise, system-wide quantification of the matrisome across the human lifespan. Comprehensive studies characterizing the matrisome repeatedly demonstrate that extracellular proteins account for a significant portion of the protein-encoding genome, subdivided rigorously into the core matrisome (structural collagens, glycoproteins, proteoglycans) and the associated matrisome (ECM regulators, secreted factors, affiliated proteins) 34.
The Proteomic Matreotype as a Biological Clock
The specific composition, relative abundance, and post-translational modification state of the matrisome in a given tissue accurately reflect its precise physiological and chronological status - a concept defined recently in the literature as the "matreotype" 34. During human aging, the matreotype undergoes highly predictable, quantifiable transitions 4.
Recent sweeping investigations utilizing the SomaScan and Olink proteomic platforms on large-scale epidemiological cohorts (such as the UK Biobank, analyzing data from 45,441 individuals) have definitively demonstrated that circulating plasma proteins can function as exceptionally accurate biological aging clocks. An analysis utilizing the Olink Explore 3072/384 platform identified over 200 specific proteins that accurately predict chronological age, achieving a remarkable Pearson correlation coefficient of r=0.94. Notably, the specific proteins contributing most heavily to the predictive power of these proteomic aging clocks are fundamentally involved in extracellular matrix interactions, systemic immune responses, and chronic inflammation 31.
Circulating Extracellular Matrix Biomarkers
Of the approximately 1,027 mapped matrisome proteins in the human genome, high-throughput analyses detect between 50% and 75% reliably within human blood plasma, allowing them to act as highly accessible systemic readouts for localized, organ-specific tissue remodeling 34. Specifically, a recent dataset successfully quantified 744 matrisome proteins in plasma, encompassing nearly 300 secreted factors, 160 ECM regulators, and 130 ECM glycoproteins 4.
Linear modeling of these plasma matrisome proteins across the human lifespan reveals a distinct, population-wide U-shaped trajectory with advancing age. Specific ECM proteins demonstrate robust positive or negative statistical associations with chronological age across multiple independent cohorts 437. An isolated ECM-based aging clock, meticulously constructed from just 14 specific plasma matrisome proteins, has demonstrated predictive accuracy comparable to massive unbiased whole-proteome clocks.
Crucially, accelerated proteomic aging - defined as instances where a patient's biological proteomic age significantly outpaces their chronological age - correlates strongly with telomere attrition, clinically defined frailty indices, and the incidence of major chronic morbidities, including Alzheimer's disease, heart failure, and diverse solid cancers 43137. Furthermore, accelerated biological aging quantified by these specific clocks is cross-sectionally associated with subclinical cerebrovascular structural changes. For instance, in the Atherosclerosis Risk in Communities (ARIC) Study, every five years of accelerated biological aging was significantly associated with larger white matter hyperintensity volumes and significantly higher odds of both lacunar and subcortical infarcts detected via 3T brain MRI 38.
| Matrisome Category | Compositional Examples | Detectability in Human Plasma | Role in Aging Biomarker Clocks |
|---|---|---|---|
| Core Matrisome: Collagens | Type I, III, IV, VII | Low to Moderate | Serve as markers of severe fibrotic degradation when fragments are detected. |
| Core Matrisome: Glycoproteins | Fibronectin, Laminins, Tenascins | High (~130 detected) | Altered circulating levels reflect loss of structural tissue mechanostasis and basement membrane decay. |
| Core Matrisome: Proteoglycans | Aggrecan, Decorin, Lumican | Moderate (~25 detected) | Indicate changes in tissue hydration and structural cushioning, particularly in osteoarthritis. |
| Associated Matrisome: Regulators | MMPs, TIMPs, LOX | High (~160 detected) | Highly predictive in 14-protein ECM clocks; reflect the active, dynamic state of matrix turnover and crosslinking. |
| Associated Matrisome: Secreted Factors | TGF-β, Wnts, Cytokines | Very High (~300 detected) | Serve as primary indicators of the Senescence-Associated Secretory Phenotype (SASP) and systemic inflammaging. |
Table 2: Breakdown of the human matrisome categories, their systemic detectability via advanced proteomics, and their specific utility as predictive biomarkers in biological aging clocks. 343137
Emerging Therapeutic Interventions and Biomaterial Innovations
Recognizing the extracellular matrix not merely as a passive victim of aging, but as a central, active pathogenic node, has heavily catalyzed the development of targeted gerotherapeutics and advanced tissue-engineered biomaterials. These innovations are explicitly designed to clear senescent cells, pharmacologically inhibit pathological crosslinking, or entirely replace irreversibly degraded matrix scaffolds.
Pharmacological Targets and Matrix Modulation
Pharmacological strategies currently in the translational pipeline are broadly divided into agents that target the direct biochemical modifications of the ECM and those that target the cellular drivers of matrix decay.
AGE Breakers and Targeted LOX Inhibitors: Because advanced glycation end-products actively drive macroscopic tissue stiffening and RAGE-mediated systemic inflammation, therapeutic compounds specifically designed to cleave existing AGE crosslinks (termed AGE breakers) or completely prevent their initial formation (AGE inhibitors) are under heavy investigation. Phytochemical screening has identified several potent natural compounds, such as extracts from Aquilaria crassna and marine red seaweed (Gigartina polycarpa), which exhibit significant anti-glycation properties and the ability to inhibit the Maillard reaction in vitro 1539. However, translating synthetic AGE breakers to the clinic has proven exceptionally difficult due to the highly complex, heterogeneous, and stable nature of in vivo AGEs 15. Concurrently, highly targeted LOX inhibitors (such as β-aminopropionitrile derivatives) are being rigorously evaluated to artificially arrest the maladaptive feed-forward loop of fibrotic enzymatic crosslinking in conditions like pulmonary hypertension and reactive myocardial fibrosis 13151620.
Dual-Function Anti-Inflammatory Agents: Pharmaceutical researchers are exploring hybrid agents engineered to mitigate both oxidative stress and the enzymatic drivers of matrix inflammation simultaneously. For example, novel dexketoprofen amide derivatives have recently been synthesized to act as specific dual-function agents. In vivo animal studies reveal that specific derivatives (e.g., derivatives 2 and 4) exhibit highly targeted COX-2 inhibition while showing exceptionally low affinity for COX-1. These compounds successfully restore systemic redox balance and heavily modulate acute inflammatory responses without inducing the severe gastrointestinal liabilities typically associated with non-selective NSAIDs 40. Additionally, modified-release formulations containing lipase and glucosidase inhibitors (such as EMP16) are currently undergoing clinical trials to target systemic metabolic drivers of aging 15.
Senotherapeutics and Proteolysis Targeting Chimeras (PROTACs)
Senolytics - specialized drugs designed to selectively induce apoptosis in senescent cells - have demonstrated substantial efficacy in clearing SASP-secreting fibroblasts and functionally restoring ECM homeostasis in preclinical models 59. First-generation senolytics, primarily repurposed anticancer agents such as Dasatinib (a broad tyrosine kinase inhibitor), Quercetin, and Fisetin (natural flavonoids), have shown significant promise in reducing the systemic senescent cell burden and restoring youthful tissue repair capacity 9. Senomorphics, including agents like rapamycin and metformin, offer an alternative approach by aiming to suppress the secretion of the SASP without eliminating the underlying cell entirely 9.
A major, persistent limitation of first-generation senolytics is their broad off-target toxicity, which poses severe clinical risks for frail, elderly patient populations. To address this critical safety bottleneck, the field of geroscience is rapidly advancing toward Proteolysis Targeting Chimeras (PROTACs). Senescence-targeting PROTACs, newly termed "SenoTACs" (including specific compounds such as ARV825, PZ15227, 753B, Gal-ARV-771, and Gal-MS99), function by actively recruiting endogenous E3 ubiquitin ligases directly to specific senescence-associated target proteins, effectively directing them for rapid proteasomal degradation 32. Because PROTACs operate in a sub-stoichiometric, purely catalytic manner, they offer vastly superior target selectivity, longer-lasting therapeutic effects, expanded target capabilities, and significantly reduced off-target toxicity compared to traditional high-dose small-molecule inhibitors 32.
Decellularized Scaffolds and Regenerative Medicine
When the native extracellular matrix is irreversibly degraded by severe age or trauma, advanced tissue engineering strategies attempt to entirely replace the structural scaffold. Decellularized extracellular matrix (dECM) has firmly emerged as a premier biomaterial in this domain 33. By subjecting allogeneic or xenogeneic donor tissues to precise chemical, physical, and enzymatic detergent protocols, the highly immunogenic cellular components (such as DNA and cell-surface antigens) are thoroughly removed. Crucially, the complex macroscopic structural architecture, MMP-sensitive cleavage peptides, and bound growth factors of the original matrix are largely preserved 33433435.
Decellularized scaffolds have demonstrated exceptionally high biocompatibility and bioactivity in vitro. They provide the necessary physical and topographical cues to directly dictate stem cell differentiation. For instance, when periodontal ligament stem cells (PDLSCs) were seeded on dECM-loaded electrospun polycaprolactone (PCL) scaffolds, the cells exhibited vastly enhanced osteogenic differentiation, increased alkaline phosphatase activity, and rapid structural mineralization compared to cells on purely synthetic scaffolds 35. Similarly, engineered anisotropic nanofibrillar scaffolds, when implanted and combined with voluntary physical exercise, significantly improved local neurovascularization and accelerated muscle regeneration in severe volumetric muscle loss models 36.
In an effort to avoid the religious restrictions, high costs, and disease transmission risks (such as prions) associated with mammalian porcine or bovine tissues, researchers are heavily exploring marine and plant-based alternatives. Decellularized tilapia skin has shown immense promise, boasting a highly favorable denaturation temperature of 68.1°C, a robust Young's modulus of 56.2 MPa, and the ability to promote high cellular metabolic activity without inducing hyperacute rejection in vivo 47. Furthermore, completely decellularized plant tissues - such as cellulose structures derived from Nopal, wild fennel, and Bougainvillea - are being utilized to provide naturally grooved, highly biocompatible biophysical cues for mammalian cell adhesion and tissue engineering, avoiding animal pathogens entirely 37.
Despite these material advancements, the translational transition from in vitro success to reliable in vivo clinical application faces considerable hurdles.
| Scaffold Modality | Primary Advantages | Key Translational Limitations |
|---|---|---|
| Mammalian Decellularized ECM (dECM) | Retains native complex architecture, bound growth factors, and highly specific mechanical cues; exceptionally bioactive. | High source variability, potential residual immunogenicity (disease transmission risk), structural degradation occurring during harsh decellularization protocols. |
| Marine/Plant Decellularized ECM | Completely bypasses mammalian disease transmission risks and religious restrictions; sustainable; highly unique topographies. | Often lacks specific mammalian integrin-binding motifs (e.g., RGD sequences in plants); requires complex biochemical functionalization to optimize mammalian cell adhesion. |
| Synthetic Polymers (e.g., PLGA, PCL) | Highly tunable porosity, degradation rates, and stiffness; highly reproducible manufacturing at commercial scale. | Complete lack of inherent bioactivity or cell-binding motifs; often degrade into toxic or acidic byproducts causing severe localized sterile inflammation. |
| Hybrid Polymeric Hydrogels (e.g., PVA, GelMA) | Combines highly tunable viscoelasticity with retained bioactive signaling molecules; injectable formats allow minimally invasive surgical delivery. | Extreme difficulty in matching the exact biomechanical stiffness of high-load musculoskeletal tissues (e.g., cartilage, Achilles tendon); struggles to maintain spatiotemporal gradients of growth factors in vivo. |
Table 3: Comprehensive comparison of tissue-engineered scaffold modalities currently utilized for the regeneration and replacement of aged or severely damaged extracellular matrices. 334334473738394041
A persistent, unresolved limitation of both dECM and hybrid synthetic scaffolds is the severe mechanical mismatch that often occurs between the engineered construct and the highly complex, dynamically loaded in vivo microenvironment 38. Furthermore, while meticulous decellularization heavily aims to eliminate complete immune rejection, trace residual DNA or subtle chemical matrix alterations induced by the processing detergents themselves can still easily elicit macrophage-mediated inflammatory responses. In clinical applications, this often leads to rapid, uncontrolled scaffold degradation or total fibrotic encapsulation by the host, rather than true, functional tissue regeneration 434042.
Extracellular matrix aging is not merely a passive structural decline, but rather a fundamental, active driver of systemic organ dysfunction. Through highly intricate mechanisms ranging from non-enzymatic glycation and pathological crosslinking to the profound disruption of cellular mechanotransduction via the YAP/TAZ and cGAS-STING pathways, the aged matrisome dictates the trajectory of age-related human disease. As the clinical understanding of dynamic ECM biology evolves, targeting the physical matrix through precision pharmacology, advanced senolytics, and highly biomimetic materials holds immense potential for drastically expanding human healthspan and successfully mitigating the biomechanical decay of aging.