Evolution of the free radical theory of aging
The Conceptual Foundations of the Free Radical Theory
Thermodynamic Realities and the Origin of Oxidative Stress
For several decades, the biological study of aging and senescence was dominated by a singular, conceptually elegant mechanism: the free radical theory of aging. Formally proposed by Denham Harman in 1956, the theory posited that chronological aging, alongside its associated degenerative diseases, is fundamentally caused by the deleterious side attacks of free radicals on vital cellular constituents 12. From a thermodynamic perspective, the vast majority of multicellular organisms rely on oxygen as a terminal electron acceptor in aerobic respiration. The redox potential of the oxygen-to-water system is highly positive, meaning that the oxidation of organic compounds by oxygen is a spontaneous and continuous thermodynamic reality 34.
During normal cellular metabolism, the reduction of oxygen generates reactive oxygen species (ROS) as unavoidable byproducts. These species include the superoxide anion, hydrogen peroxide, and the highly volatile hydroxyl radical. The original premise of Harman's theory argued that these reactive molecules indiscriminately damage biomolecules 56. Over time, the theory hypothesized that the inability of endogenous antioxidant defense systems - such as superoxide dismutase, catalase, and glutathione peroxidase - to perfectly neutralize all ROS leads to a progressive accumulation of macromolecular damage. This time-dependent accumulation of cellular damage was viewed as the primary driver of the macroscopic aging phenotype, manifesting ultimately as tissue degradation, loss of physiological integrity, and increased vulnerability to death 78.
For much of the late twentieth century, the free radical theory provided a cohesive molecular rationale for why organisms age. It aligned with broader evolutionary concepts, notably the "disposable soma" theory, which suggests that organisms invest their finite metabolic resources in reproduction and early-life survival rather than perpetual somatic maintenance and flawless DNA repair 9. Because the force of natural selection weakens precipitously with advancing chronological age - after an organism has passed its reproductive peak - evolutionary mechanisms fail to select against the late-life accumulation of oxidative damage 910.
The Mitochondrial Iteration of the Free Radical Theory
As cellular and molecular biology advanced, Harman's original proposition was refined into the mitochondrial free radical theory of aging (MFRTA). Mitochondria are the primary sites of oxidative phosphorylation and, consequently, the main endogenous source of ROS in eukaryotic cells. It is estimated that a fraction of the total oxygen consumed by resting cells is converted into ROS during ATP production, making mitochondria the most prolific generators of oxidative stress 46.
The MFRTA suggested a vicious, positive-feedback cycle of cellular degradation. According to this model, ROS produced by the electron transport chain leak into the mitochondrial matrix and cause oxidative damage primarily to mitochondrial DNA (mtDNA), which resides in close proximity to the site of ROS generation. Because mtDNA lacks the protective histone proteins and robust repair mechanisms characteristic of nuclear DNA, it is uniquely susceptible to mutagenic damage 711.
The paradigm argued that oxidative damage to mtDNA results in the synthesis of defective mitochondrial proteins, which are subsequently incorporated into the electron transport chain. This defect leads to further uncoupling of the chain, causing a progressive decline in efficient ATP production and an exponential increase in ROS leakage 67. This accelerating cycle of mitochondrial dysfunction and global cellular damage was widely accepted as the fundamental biological clock of aging. Observational studies documenting age-associated increases in oxidative lesions, such as 7,8-dihydro-8-oxo-deoxyguanosine (8-oxodG), heavily bolstered the view that oxidative damage was the primary etiology of cellular senescence and organismal mortality 2.
Empirical Challenges and the Decline of the Free Radical Paradigm
Discrepancies in Genetic and Animal Models
If the free radical theory of aging were a universal biological law, manipulating the balance of oxidants and antioxidants should predictably and consistently alter maximum lifespan. Specifically, administering exogenous antioxidants or genetically overexpressing endogenous antioxidant enzymes should extend lifespan, while disabling antioxidant defenses to increase oxidative stress should accelerate aging and reduce survival.
However, experimental data from invertebrate and mammalian models consistently failed to support these hypotheses. Studies overexpressing antioxidant enzymes such as superoxide dismutase (SOD) or catalase in mice repeatedly failed to demonstrate significant lifespan extension or global improvements in healthspan 4. Conversely, genetic knockout models that disabled antioxidant defenses - resulting in verifiably elevated levels of oxidative stress - often resulted in increased specific pathologies but did not necessarily shorten maximum lifespan or accelerate the fundamental, systemic aging process in a uniform manner 4.
Furthermore, comparative biology presented glaring contradictions to the theory. Certain exceptionally long-lived species, such as the naked mole-rat, exhibit high levels of oxidative damage from a very young age. Despite this chronic oxidative stress, these organisms maintain remarkable longevity and resistance to age-related diseases, directly contravening the core assumption that cumulative oxidative damage strictly dictates maximum lifespan 24.
Deeper investigations into mitochondrial genetics further complicated the MFRTA. Advanced sequencing techniques indicated that age-associated mtDNA mutations are frequently transition mutations resulting from inherent replication errors made by the mitochondrial polymerase gamma, rather than direct transversions caused by oxidative lesions 2. Furthermore, longitudinal evidence suggests that normal mtDNA turnover leads to the clonal expansion of pre-existing mutations rather than a gradual, random accumulation of new oxidative damage over time 2. These nuances began to reveal profound structural flaws in the foundation of the free radical theory, indicating that while mitochondrial dysfunction is undeniably central to aging, the simple linear model of ROS-induced damage was fundamentally incomplete.
The Clinical Rejection of Dietary Antioxidant Supplementation
The most definitive and highly publicized evidence challenging the free radical theory emerged from large-scale human clinical trials. These trials were designed to test the logical conclusion of the theory: if oxidative damage causes aging and disease, then supplementing the human diet with high doses of antioxidant vitamins should prevent cancer, cardiovascular disease, and all-cause mortality.
Instead of proving the theory, the results of these randomized controlled trials were overwhelmingly null, and in several high-profile cases, actively harmful to human health. Four landmark trials fundamentally shifted the scientific and medical consensus away from antioxidant supplementation:
The Beta-Carotene and Retinol Efficacy Trial (CARET) was initiated to test whether daily beta-carotene (30 mg) and retinyl palmitate (25,000 IU) could prevent lung cancer in high-risk populations, specifically heavy smokers and asbestos workers. The trial, involving over 18,000 participants, was halted prematurely in January 1996 1213. Investigators discovered a shocking 28% increase in the incidence of lung cancer, a 17% increase in all-cause mortality, and a higher rate of cardiovascular disease mortality among participants receiving the active antioxidant supplements compared to the placebo group 1213. Extended post-intervention follow-up revealed that the elevated relative risks for lung cancer and all-cause mortality persisted long after the supplementation had ended, with female smokers exhibiting particularly elevated post-trial mortality risks 12.
Concurrently, the Alpha-Tocopherol, Beta-Carotene Cancer Prevention (ATBC) Study was conducted in Finland among 29,000 male smokers. Similar to CARET, this trial found that daily beta-carotene supplementation increased lung cancer incidence by 18% and overall mortality by 8% 1314. The cohort receiving alpha-tocopherol (vitamin E) saw no reduction in lung cancer incidence and, alarmingly, experienced a 20% increased risk of dying from prostate cancer during post-trial follow-up 1415.
The Selenium and Vitamin E Cancer Prevention Trial (SELECT) represented one of the largest cancer prevention trials ever undertaken. It randomized over 35,000 healthy men to receive high-dose vitamin E (400 IU), selenium (200 micrograms as l-selenomethionine), a combination of both, or a placebo to evaluate prostate cancer prevention 1617. The rationale was based on earlier, smaller trials like the Nutritional Prevention of Cancer (NPC) trial, which suggested selenium might reduce cancer risks in populations with low baseline selenium levels 16. However, SELECT was discontinued after a median of 5.46 years due to complete futility 16. Subsequent follow-up data analysis revealed a statistically significant 17% increased risk of prostate cancer in the vitamin E-only arm 1718. High-dose selenium supplementation failed to yield cardiovascular or survival benefits, and subsequent long-term analyses from independent cohorts (such as the PRECISE study) linked high-dose selenium (300 micrograms/day) to an increased risk of all-cause mortality over a 10-year follow-up period 19.
Finally, the Physicians' Health Study II evaluated vitamins E and C in the prevention of cardiovascular disease and cancer among 14,641 male physicians. Over a decade of follow-up, neither vitamin E nor vitamin C, alone or in combination, reduced the risk of cancer, cardiovascular events, or all-cause mortality 1820.
Meta-Analyses on Antioxidant Interventions and Mortality
The compounding negative results from these individual mega-trials prompted exhaustive, systematic meta-analyses to settle the debate. A comprehensive Cochrane systematic review evaluating 78 randomized clinical trials encompassing nearly 300,000 participants assessed the impact of beta-carotene, vitamin A, vitamin C, vitamin E, and selenium on mortality 2122.
The meta-analysis revealed that antioxidant supplements had no significant protective effect on mortality across the general population. More critically, when the analysis was restricted to the 56 trials with the highest methodological quality (low risk of bias), antioxidant supplementation was associated with a statistically significant 4% increase in all-cause mortality (Relative Risk 1.04; 95% CI 1.01 to 1.07) 2122. Further subgroup analyses indicated that beta-carotene, vitamin E, and vitamin A were individually associated with increased mortality risks, while vitamin C and selenium showed no significant effect on extending lifespan 2122.
| Clinical Trial | Target Population | Interventions Tested | Primary Findings Regarding Health and Mortality |
|---|---|---|---|
| CARET | High-risk (smokers, asbestos workers) | Beta-carotene, Retinyl palmitate | 28% increase in lung cancer; 17% increase in all-cause mortality. Trial halted prematurely. 1213 |
| ATBC | Male smokers in Finland | Alpha-tocopherol, Beta-carotene | 8% higher all-cause mortality; increased lung cancer with beta-carotene; 20% increased prostate cancer mortality with vitamin E. 1415 |
| SELECT | Healthy males >50 years old | Vitamin E, Selenium (l-selenomethionine) | No cancer prevention benefit. Significant 17% increased risk of prostate cancer with vitamin E. 161718 |
| Physicians' Health Study II | Male physicians >50 years old | Vitamin E, Vitamin C | No reduction in cancer risk, cardiovascular disease, or all-cause mortality with single or combined supplementation. 1820 |
| PRECISE | Volunteers 60 - 74 years old | Selenium (100, 200, 300 μg/d) | 300 μg/d dose associated with an increased hazard ratio for all-cause mortality after 10-year follow-up. 19 |
While some isolated secondary prevention trials or studies conducted in heavily malnourished populations showed marginal, context-specific benefits, the overarching clinical conclusion was definitive: broad, high-dose antioxidant supplementation does not extend human lifespan and actively disrupts critical physiological homeostasis 2123. These findings served as the empirical death knell for the simplistic interpretation of the free radical theory, forcing the field of biogerontology to search for entirely new paradigms to explain the biological mechanics of aging 1124.
The Paradoxical Growth of the Commercial Antioxidant Market
Despite the overwhelming scientific consensus that exogenous antioxidant supplementation fails to extend human lifespan or meaningfully mitigate age-related diseases, a stark disconnect exists between clinical evidence and consumer behavior. The global market for dietary antioxidant supplements continues to experience robust, uninterrupted growth.
Market analysis data indicates that the global antioxidant vitamin market was valued at approximately USD 1.17 billion in 2024 and is projected to reach USD 2.00 billion by 2032, expanding at a compound annual growth rate (CAGR) of roughly 6.9% 25. Broader definitions of the market, encompassing "super antioxidants" and specialized dietary functional foods, place the market valuation much higher. Some analyses project the global antioxidant supplement market to grow from USD 4.8 billion in 2025 to USD 9.1 billion by 2035 26.
This market expansion is driven by deeply entrenched public perceptions that equate oxidative stress exclusively with toxic damage and aging. Consumer demand is fueled by an increasing focus on preventive healthcare, immune resilience, and the pursuit of clean-label, plant-based nutraceuticals 252728.
| Market Segment Definition | Estimated Base Year Value | Projected Future Value | Estimated CAGR | Key Growth Drivers |
|---|---|---|---|---|
| Antioxidant Vitamins | USD 1.17 Billion (2024) | USD 2.00 Billion (2032) | 6.9% | Preventive healthcare trends, clean-label plant-based ingredients, cosmetic applications. 25 |
| Super Antioxidant Supplements | USD 588.3 Million (2025) | USD 995.5 Million (2035) | 5.3% | Immune boosting demand post-pandemic, Pycnogenol and CoQ10 popularity. 29 |
| Global Antioxidant Supplements (Broad) | USD 4.8 Billion (2025) | USD 9.1 Billion (2035) | 6.7% | Rising disposable income, proactive health management, anti-aging aesthetics. 26 |
Geographically, North America currently holds the largest market share, driven by aggressive marketing of anti-aging products and high disposable income. However, the Asia-Pacific region is projected to experience the fastest growth, supported by rapid urbanization and the integration of traditional medicine with modern functional food trends 2527. The commercial success of these products highlights the profound difficulty in translating complex biological paradigm shifts - such as the rejection of the free radical theory - into actionable public health knowledge.
The Discovery of Mitohormesis and Reactive Oxygen Species Signaling
Redefining Reactive Oxygen Species as Cellular Messengers
The comprehensive failure of the free radical theory necessitated a profound reevaluation of the biological role of ROS within the cell. If ROS were purely destructive metabolic byproducts as Harman originally envisioned, neutralizing them with high-dose antioxidants should have yielded unequivocal clinical benefits. The discovery that antioxidant supplementation could be clinically detrimental led researchers to the realization that ROS are not merely toxic waste; they are critical, highly conserved signaling molecules essential for cellular survival, adaptation, and stress resilience 430.
This paradigm shift gave rise to the concept of mitochondrial hormesis, commonly referred to as "mitohormesis." Hormesis is a widely recognized biological phenomenon characterized by a biphasic dose-response relationship.
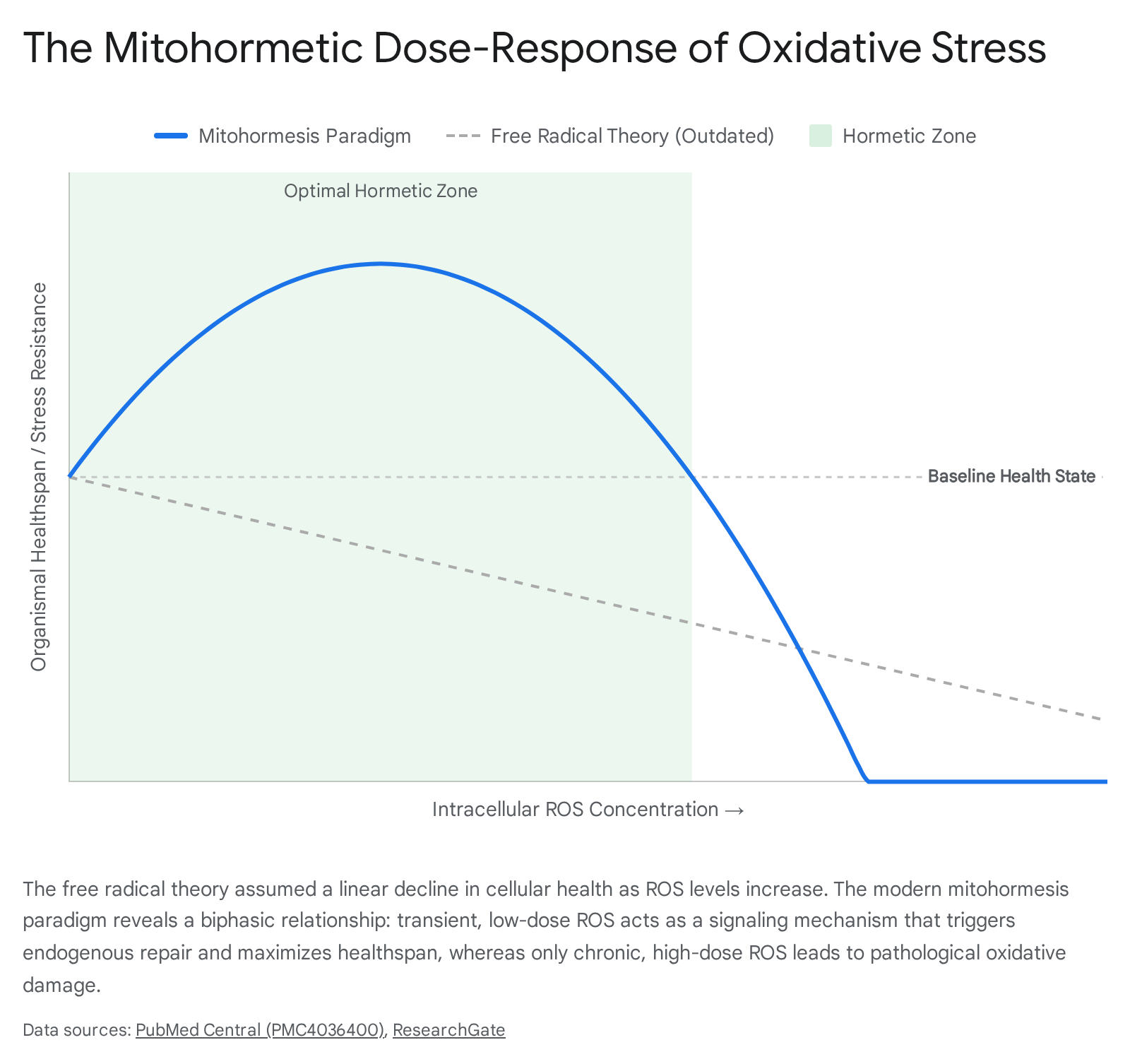
Under a hormetic framework, low-dose exposure to a stressor induces an adaptive, beneficial response that strengthens the organism, while high-dose exposure to the exact same stressor results in toxicity, damage, and death 431.
Under the mitohormesis model, transient and low-level increases in mitochondrial ROS - such as those naturally generated during physical exercise, caloric restriction, or mild hypoxia - act as vital signaling messengers 43132. These localized bursts of ROS communicate physiological stress states from the mitochondria to the nucleus, triggering the activation of robust systemic defense mechanisms.
For example, mild oxidative stress facilitates the dissociation of the transcription factor Nrf2 from its cytoplasmic repressor Keap1. Once liberated, Nrf2 translocates to the nucleus, binding to Antioxidant Response Elements (AREs) across the genome to significantly upregulate the endogenous production of antioxidant enzymes, phase II detoxification proteins, and vital cellular repair machinery 4.
By initiating this "vaccination-like" adaptive response, low-level ROS actually prime the cell to resist subsequent, more severe metabolic or oxidative insults. This process ultimately enhances stress resistance, improves metabolic flexibility, and extends lifespan in experimental models 43133. This elegant feedback loop explains precisely why the chronic administration of high-dose exogenous antioxidants is counterproductive: by prematurely quenching these vital ROS signals, artificial antioxidant supplements prevent the activation of the body's endogenous defense pathways, leaving the organism functionally weaker and more vulnerable to age-related decline 4.
The Intersection of Mitohormesis, AMPK, and mTOR Pathways
The discovery and validation of mitohormesis aligned perfectly with simultaneous, groundbreaking advancements in understanding the cellular nutrient-sensing longevity pathways, primarily the AMP-activated protein kinase (AMPK) and the mechanistic target of rapamycin (mTOR) pathways. Aging is highly dependent on cellular energy status, and these two pathways serve as the master regulators of systemic metabolic homeostasis.
AMPK is a critical intracellular energy sensor that is activated in response to energy deficits 3435. The exact mechanism by which ROS activates AMPK was historically debated in the literature. Initial hypotheses suggested that ROS directly oxidized redox-sensitive cysteine residues (specifically Cys-299 and Cys-304) on the AMPK alpha subunit to trigger structural activation 343536. However, robust cellular re-investigations revealed that substituting these putative redox-active cysteines with alanines did not prevent AMPK activation in the presence of hydrogen peroxide 3536.
Instead, the modern consensus indicates that AMPK responds to ROS indirectly. Moderate oxidative stress mildly inhibits the mitochondrial respiratory chain, causing a subtle decline in the rate of ATP production and a corresponding increase in the cellular AMP:ATP and ADP:ATP ratios 34353637. This energetic shift relies on the canonical adenine nucleotide-dependent activation of the AMPK gamma subunit, meaning AMPK is fundamentally an energy sensor responding to ROS-induced metabolic shifts rather than a direct redox sensor 3537.
Once activated, AMPK serves a dual role: it inhibits energy-consuming anabolic processes and forcefully stimulates energy-generating catabolic pathways, including glucose uptake, lipid oxidation, and mitochondrial biogenesis 3538. Crucially, AMPK directly phosphorylates and inhibits the mTOR complex (specifically mTORC1). While mTOR is absolutely vital for cell growth, protein synthesis, and development when nutrients are abundant, the chronic hyperactivation of mTOR accelerates the aging process by promoting cellular senescence, exhausting limited stem cell pools, and inhibiting cellular recycling mechanisms 353940.
Inhibiting mTOR - either indirectly via AMPK activation, behaviorally through caloric restriction, or pharmacologically with agents like rapamycin - upregulates macroautophagy. Autophagy allows the cell to systematically degrade and clear misfolded proteins and dysfunctional organelles, renewing the cellular environment 394041. Consequently, the mild ROS generated during stress conditions acts as the initial molecular trigger that initiates the AMPK/mTOR regulatory cascade, shifting the cell from a state of rapid growth to a state of robust somatic maintenance and repair. Intervening with powerful antioxidants prematurely terminates this precise cascade, providing a complete molecular explanation for the ultimate failure of the free radical theory 4.
The Hallmarks of Aging as the Modern Conceptual Framework
Transition to a Pluralistic and Interconnected Model
The realization that aging could not be distilled down to a single mechanism like free radical damage led the biological community to seek a vastly more comprehensive and systems-level framework. In 2013, Carlos López-Otín and an international consortium of colleagues published a seminal paper in the journal Cell outlining "The Hallmarks of Aging." This paper successfully synthesized decades of disparate research into nine distinct but deeply interconnected cellular and molecular denominators of the aging process 7842. In 2023, reflecting a decade of rapid advancement and massive data accumulation in geroscience, the framework was officially expanded to encompass twelve hallmarks 434544.
To qualify as a genuine hallmark of aging, a biological process must strictly fulfill three empirical criteria: it must manifest progressively during normal physiological aging, its experimental aggravation must demonstrably accelerate the aging process, and its experimental amelioration must decelerate the aging process and extend healthy lifespan 84243. The modern framework recognizes that aging is the cumulative, systemic result of a complex, multilayered network of deteriorative processes rather than a single toxic vector. These twelve hallmarks are hierarchically organized into three distinct operational categories: Primary, Antagonistic, and Integrative hallmarks 424445.
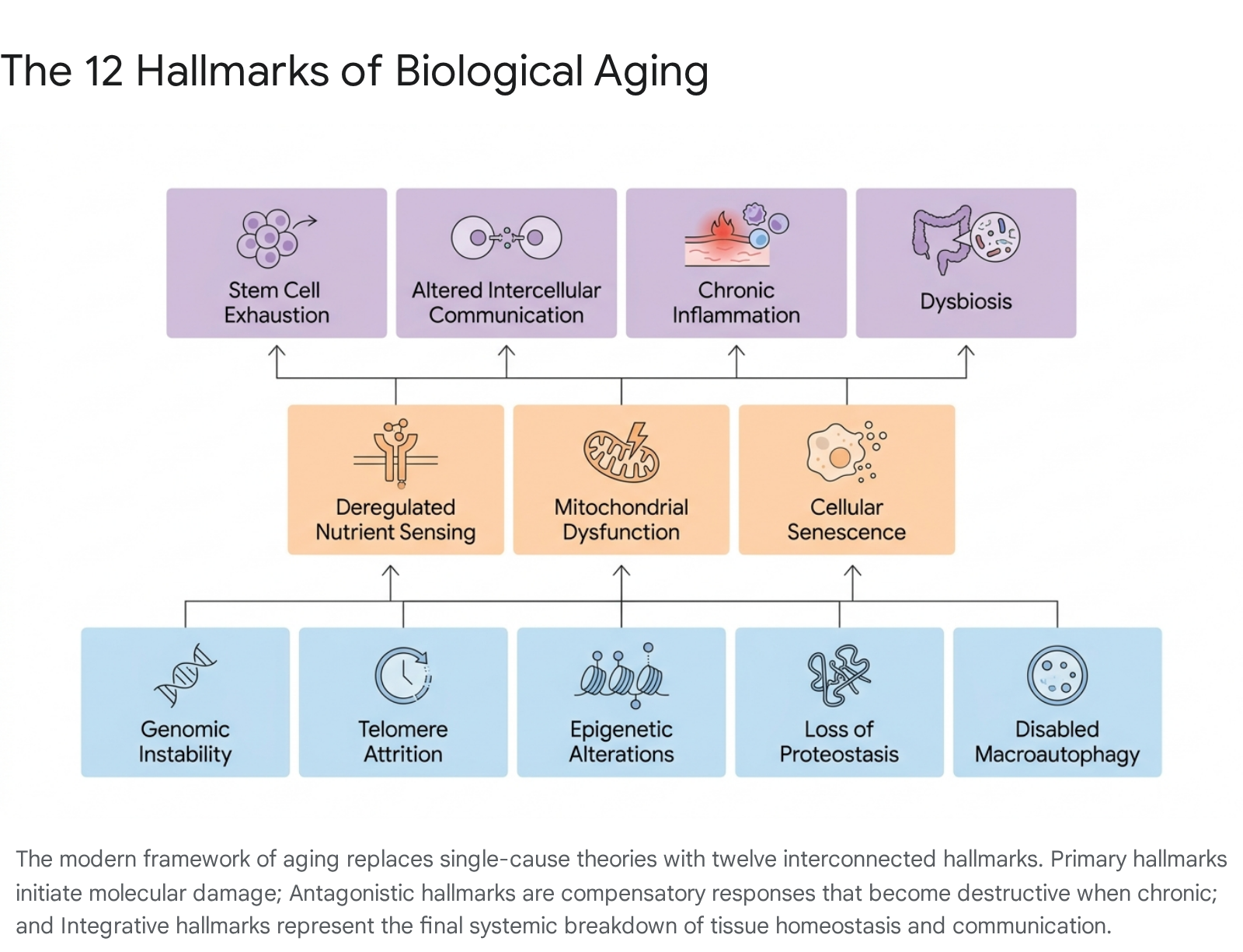
Primary Determinants of Cellular Aging
The primary hallmarks represent the fundamental, initiating causes of cellular aging. These are the underlying molecular damages that inevitably accumulate over time due to intrinsic thermodynamic instability and routine environmental insults 4244. They act "from the inside" of the cell and are universally negative in their biological impact:
The first primary hallmark is genomic instability. Over the course of an organism's life, DNA is constantly subjected to endogenous replication errors and exogenous insults, such as UV radiation and chemical mutagens. Despite the existence of robust DNA repair mechanisms - including base excision repair, nucleotide excision repair, and homologous recombination - damage slowly and inevitably accumulates. This damage manifests as point mutations, chromosomal alterations, and deletions, which fundamentally corrupt the instructions required for cellular function 7424647.
Telomere attrition represents the second primary hallmark. Telomeres are the protective, repetitive nucleotide sequences capping the ends of linear chromosomes. Because standard DNA polymerases cannot fully replicate the extreme terminal ends of DNA strands, telomeres shorten slightly with each cell division. When telomeres reach a critically short length, they lose their protective capacity and trigger a persistent DNA damage response. This evolutionary safeguard prevents genomic chaos but forcefully drives the cell into replicative senescence or apoptosis, limiting tissue renewal 746.
Epigenetic alterations constitute the third hallmark. Aging involves profound and widespread changes to the epigenome, including DNA methylation drift, post-translational histone modifications, and systemic chromatin remodeling. These alterations distort youthful, precise gene expression profiles. The epigenetic drift can turn off critical repair and tumor-suppressor genes while aberrantly activating inflammatory or oncogenic pathways, degrading cellular identity 746.
The fourth primary hallmark is the loss of proteostasis. The failure of protein homeostasis involves a gradual decline in the cell's sophisticated machinery required to fold proteins correctly, traffic them accurately, and degrade damaged or obsolete ones. This failure leads to the toxic accumulation of misfolded protein aggregates within the cell, a hallmark driver of severe age-related neurodegenerative diseases such as Alzheimer's and Parkinson's 74245.
Finally, disabled macroautophagy was formally added to the primary tier in the 2023 update. This hallmark specifically denotes the age-related decline in the cell's primary bulk recycling system. A failure in macroautophagy prevents the clearance of damaged organelles - such as senescent mitochondria - and large protein aggregates. This deficit chokes cellular metabolism and aggressively compounds the loss of proteostasis 424344.
Antagonistic Responses and the Recontextualization of Oxidative Stress
The antagonistic hallmarks represent a critical conceptual leap in aging biology. These are biological responses that originally evolved to protect the organism from the damage caused by primary hallmarks or transient environmental stress. At low intensities or during youth, these responses are highly beneficial. However, when these responses become chronically activated or exacerbated in old age, they subvert their original purpose and become actively deleterious, driving the macroscopic aging phenotype 424448.
It is specifically within this category that the remnants of the free radical theory now reside, having been radically recontextualized as the hallmark of mitochondrial dysfunction. Mitochondria lose their bioenergetic efficiency with advancing age, leading to decreased ATP production and increased ROS leakage. However, rather than viewing ROS solely as destructive agents, mitochondrial dysfunction is now properly understood to involve a systemic failure of mitohormesis 4244. In youth, mild mitochondrial stress triggers protective adaptation. In aging, the accumulation of primary damage overwhelms this adaptive system, leading to sustained ROS production that causes membrane permeabilization, triggers the inflammasome, and induces cell death 64251. The hallmark is defined by the loss of the delicate energetic and signaling balance, not merely the presence of free radicals.
Deregulated nutrient sensing is the second antagonistic hallmark. The nutrient-sensing network - which includes the insulin/IGF-1 pathway, mTOR, AMPK, and sirtuins - evolved to tightly coordinate cellular growth with environmental nutrient availability. While high mTOR activity drives necessary and beneficial growth in youth, its failure to downregulate in adulthood results in unchecked anabolism, systematically suppressing autophagy and driving metabolic diseases like obesity and type 2 diabetes. Moderating these pathways via caloric restriction or pharmacological inhibitors consistently extends lifespan across diverse model organisms 42454851.
Cellular senescence is the third antagonistic hallmark. Senescence is a state of irreversible cell-cycle arrest triggered by unresolvable damage, such as critical telomere shortening or severe oxidative stress. Initially, senescence serves as a highly beneficial, evolutionary tumor-suppressive mechanism that prevents damaged, potentially malignant cells from proliferating 4246. It also aids in tissue repair and wound healing. However, the immune system's ability to locate and clear these "zombie cells" declines precipitously with age. Consequently, senescent cells accumulate in tissues and secrete a highly toxic mix of inflammatory cytokines, proteases, and growth factors known as the Senescence-Associated Secretory Phenotype (SASP). The SASP degrades surrounding tissue architecture and triggers secondary senescence in neighboring healthy cells, acting as a primary engine of tissue aging 454851.
Integrative Consequences of Tissue and Systemic Decline
The integrative hallmarks emerge strictly as the downstream consequences of the accumulated damage from primary mechanisms and the failed, chronic compensatory responses of the antagonistic mechanisms. These hallmarks act at the macroscopic tissue and systemic levels, leading directly to the clinical manifestations of aging 424445:
Stem cell exhaustion is a critical integrative hallmark. The regenerative capacity of tissues dramatically declines as somatic stem cell pools are depleted or lose their functional pluripotency due to accumulated DNA damage, senescence, and epigenetic drift. This exhaustion fundamentally prevents the ongoing repair and maintenance of highly proliferative tissues, including muscle, bone, skin, and the hematopoietic immune system 74245.
Altered intercellular communication reflects the reality that aging disrupts the complex, finely tuned signaling networks between cells, altering endocrine, neuroendocrine, and neuronal pathways. This systemic breakdown leads to widespread hormonal imbalances and a failure to coordinate physiological responses to stress 745.
Chronic inflammation, frequently termed "inflammaging," was added in 2023 to acknowledge that aging is characterized by a low-grade, sterile, and chronic systemic inflammatory state. Driven continuously by the SASP from senescent cells, accumulating cellular debris from disabled autophagy, and a declining, misdirected immune system, inflammaging aggressively accelerates tissue scarring, neurodegeneration, and cardiovascular disease 43454546.
Dysbiosis, the final integrative hallmark added in 2023, refers to the age-related alteration of the host microbiome, particularly within the gastrointestinal tract. Shifts in microbial populations compromise intestinal barrier integrity, alter systemic nutrient absorption, and exude pro-inflammatory metabolites directly into the bloodstream, deeply affecting both systemic metabolic regulation and immune health 434545.
Current Consensus on Evolutionary Aging and Therapeutic Horizons
Synthesis of Evolutionary Theories and Molecular Hallmarks
The transition from the simplistic free radical theory to the highly nuanced hallmarks of aging reflects a profound maturation of the field that finally bridges cellular molecular biology with macroscopic evolutionary theory. In 2024, the International Association of Gerontology and Geriatrics (IAGG) highlighted a growing, unified consensus that aging must be viewed as an emergent, non-adaptive byproduct of evolution rather than a programmed self-destruct sequence 1052.
Classical evolutionary frameworks - specifically mutation accumulation, antagonistic pleiotropy, and the disposable soma theory - posit that aging occurs because the force of natural selection declines monotonically with age 910. Because predators, disease, and environmental hazards ensure that very few organisms survive to old age in wild ecologies, evolutionary processes do not prioritize or select for the genetic programs necessary for infinite, flawless molecular repair 9.
The antagonistic hallmarks, particularly deregulated nutrient sensing and cellular senescence, perfectly mirror the evolutionary concept of antagonistic pleiotropy. This concept dictates that genetic traits that are highly beneficial for rapid growth, reproduction, and robust cancer suppression early in life become inherently destructive when an organism happens to survive beyond its evolutionary warranty into old age 10424849. The updated hallmarks model successfully integrates these evolutionary realities, acknowledging that aging is the gradual, systemic collapse of biological maintenance, rather than a single toxic variable like oxidation running unchecked 104250.
Implications for Therapeutics and Lifespan Extension
The decisive downfall of the free radical theory fundamentally altered the trajectory of biopharmaceutical research and drug development. The realization that exogenous antioxidant vitamins generally fail to influence human longevity - and may even blunt beneficial stress adaptations and exercise responses - has redirected global research efforts toward precise interventions that target the underlying cellular hallmarks 45551.
Modern longevity research now relies heavily on modulating the precise nutrient-sensing networks that govern the antagonistic hallmarks. Rather than attempting to neutralize ROS directly, novel interventions aim to trigger the downstream repair pathways that ROS naturally activate during mitohormesis. Molecules that activate AMPK (such as metformin and berberine) or inhibit mTOR (such as rapamycin) effectively trick the cell into a physiological state of perceived nutrient scarcity. This artificial signal upregulates macroautophagy, improves mitochondrial dynamics, and bolsters endogenous DNA repair mechanisms without requiring the cell to undergo actual starvation 353941. The therapeutic objective is to maintain metabolic flexibility by cyclically activating these pathways, perfectly mimicking the evolutionary stress signals that maintain youthful homeostasis 41.
The success of targeting metabolic and nutrient-sensing pathways over antioxidant therapy is increasingly evident in major clinical trials. For instance, the recent Semaglutide Effects on Heart Disease and Stroke in Patients With Overweight or Obesity (SELECT) trial demonstrated that using a GLP-1 receptor agonist to address metabolic dysfunction yielded a profound 20% reduction in the risk of composite cardiovascular endpoints and a significant reduction in all-cause mortality, underscoring that metabolic regulation is a vastly more effective lever for healthspan extension than attempting to alter cellular redox states 525853.
Furthermore, researchers are aggressively targeting cellular senescence through the development of "senolytics." These compounds - such as the combination of dasatinib and quercetin - are designed to selectively induce apoptosis in senescent cells, thereby clearing the toxic SASP from tissues and directly alleviating the chronic inflammation characteristic of inflammaging 46. Restoring youthful epigenetic landscapes via partial cellular reprogramming (utilizing the Yamanaka transcription factors) is also being heavily explored as a method to reverse epigenetic drift and rescue stem cell exhaustion at a systemic level 244246.
Ultimately, the fall of the free radical theory of aging was not a failure of scientific inquiry, but a necessary, dialectical stepping stone. It provided the foundational understanding of macromolecular damage that eventually evolved into the sophisticated, systems-biology approach defined by the hallmarks of aging. By shifting focus from the futile attempt to eradicate oxidative stress to the nuanced, targeted management of interconnected cellular networks, the field of geroscience is now infinitely better positioned to develop therapeutic interventions that may meaningfully extend human healthspan and longevity.