Endocrine functions of bone and osteocalcin
Introduction to Skeletal Endocrinology
The biological understanding of the vertebrate skeleton has undergone a profound paradigm shift over the past two decades. Historically, bone was viewed almost exclusively as a static structural scaffold designed for locomotion, the protection of internal organs, the housing of the hematopoietic niche, and serving as an inert reservoir for systemic calcium and phosphate homeostasis 123. Within this traditional framework, the physiological regulation of bone was understood through a unilateral and mechanical lens. The skeleton was primarily considered a target organ that responded passively to mechanical loading and systemic endocrine signals, including parathyroid hormone (PTH), calcitonin, sex steroids, and vitamin D derivatives 23.
Recent discoveries have fundamentally dismantled this structural-centric view, revealing that bone acts as a highly dynamic, pleiotropic endocrine organ 134. The skeleton actively secretes highly specialized, bone-derived factors - frequently termed osteokines - that exert systemic regulatory effects on multiple distant organs. These target tissues include the pancreas, adipose tissue, skeletal muscle, brain, and cardiovascular system 124. The primary molecular mediators of this skeletal endocrine network include osteocalcin (OCN), fibroblast growth factor 23 (FGF23), and lipocalin-2 (LCN2) 134.
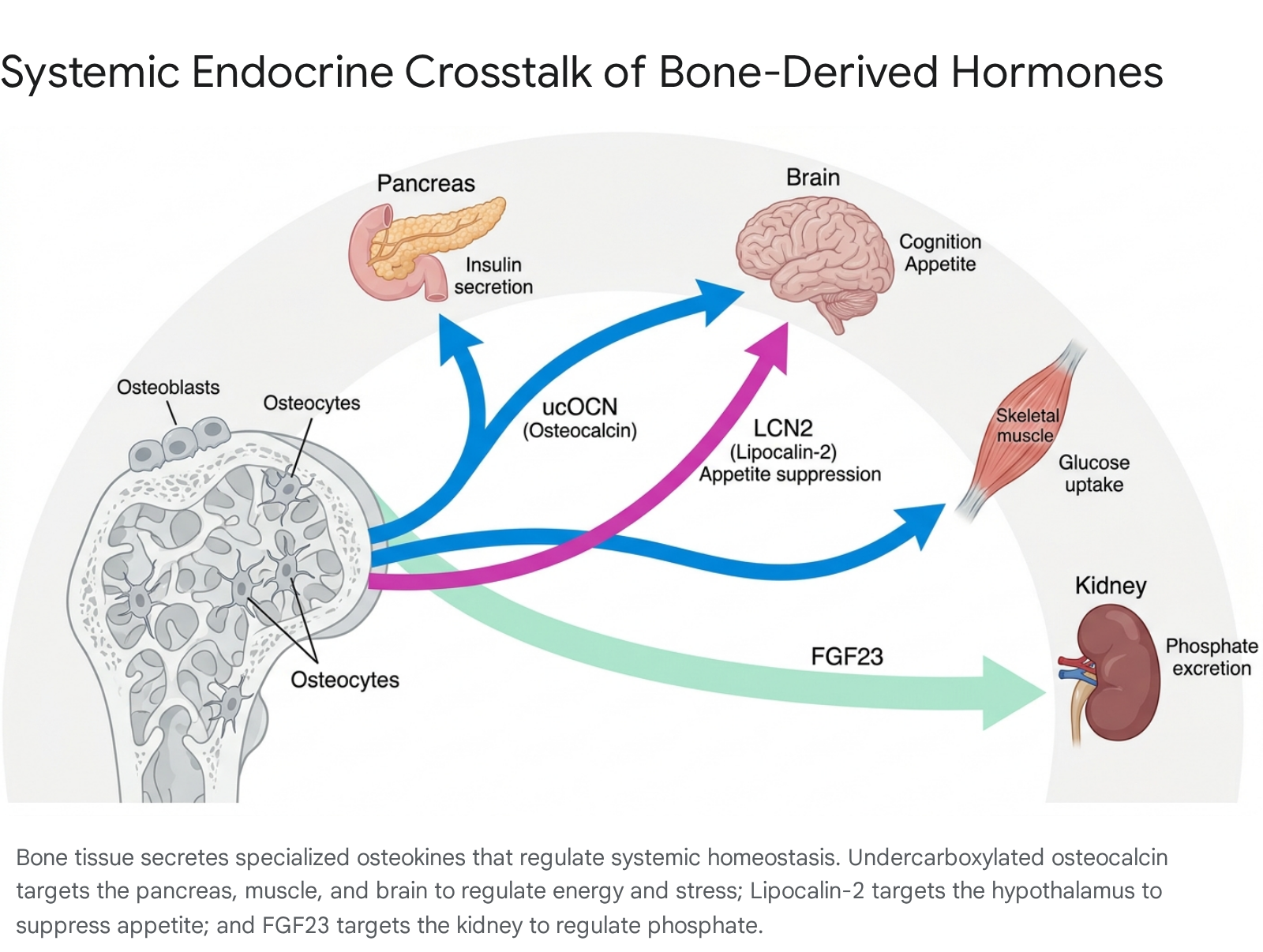
The recognition and subsequent characterization of these molecules have repositioned the skeleton as a central integrating node for whole-body energy metabolism, acute stress physiology, and nutritional homeostasis.
Molecular Biology and Biosynthesis of Osteocalcin
Genomic Structure and Protein Translation
Osteocalcin, alternatively referred to as bone gamma-carboxyglutamate protein, is a small, highly conserved peptide consisting of 49 amino acids in humans and 46 amino acids in mice 135. It is encoded by the BGLAP gene and represents the most abundant non-collagenous protein found within the bone extracellular matrix 135. Osteocalcin is synthesized almost exclusively by mature osteoblasts during the late stages of cellular differentiation 367.
The biosynthetic pathway begins with the translation of a precursor molecule, pre-pro-osteocalcin, which is subsequently transported into the endoplasmic reticulum and enzymatically cleaved to form pro-osteocalcin 367. Following this cleavage, the protein undergoes a critical post-translational modification that dictates its physiological function, allowing it to transition between a structural matrix protein and a systemic signaling hormone.
Vitamin K-Dependent Carboxylation and Matrix Integration
The defining biochemical feature of osteocalcin is its post-translational carboxylation. Within the endoplasmic reticulum of the osteoblast, specific glutamic acid (Glu) residues - located at positions 17, 21, and 24 - undergo $\gamma$-carboxylation 368. This process is catalyzed by the enzyme $\gamma$-glutamyl carboxylase (GGCX), a reaction that is strictly dependent on the presence of Vitamin K (primarily in the MK-4 and MK-7 menaquinone forms), which serves as an essential electron-donating cofactor 369.
The addition of these carboxyl groups generates fully carboxylated osteocalcin (cOCN), also referred to as 3 Gla-OCN. The $\gamma$-carboxyglutamic acid residues confer a highly negative charge to the protein, granting it a profound binding affinity for positively charged calcium ions 71011. This high-affinity binding permits cOCN to integrate directly into the bone matrix, where it binds strongly to the (001) and (010) crystallographic facets of hydroxyapatite crystals 10.
Molecular simulations and rigorous multiscale structural analyses have demonstrated that cOCN plays a critical role in bone mineralization. It guides the formation and integration of hydroxyapatite, ensuring that the mineral crystallites align parallel to the surrounding collagen fibrils 101213. This precise structural orchestration is indispensable for optimizing bone material quality. It dictates mineral crystal size, limits abnormal calcification, and provides the necessary mechanical strength and toughness to resist fracture, a property that is highly independent of gross bone mass or bone mineral density (BMD) measurements 81213.
Osteoclast-Mediated Decarboxylation and Endocrine Activation
Osteocalcin's transition from a structural component to a circulating systemic hormone is tightly coupled to the physiological process of bone resorption 168. The extracellular bone matrix functions as an enormous biological repository for carboxylated osteocalcin. When bone remodeling is required, multinucleated osteoclasts attach to the bone surface, forming a sealed resorption lacuna. The osteoclasts then secrete hydrochloric acid via vacuolar-type H+-ATPases, drastically lowering the pH in the local microenvironment to approximately 4.5 1810.
This highly acidic microenvironment triggers two simultaneous chemical events. First, it dissolves the hydroxyapatite mineral, liberating the entrapped matrix-bound proteins 110. Second, the low pH physically alters the molecular conformation of osteocalcin, driving an active chemical decarboxylation that strips the carboxyl groups from the three glutamic acid residues 16. This reaction generates undercarboxylated osteocalcin (ucOCN, also referred to as Glu-OCN or 3 Glu-OCN) 111.
Lacking the carboxyl groups, ucOCN loses its affinity for calcium and can no longer rebind to bone mineral 111. Consequently, the unbound ucOCN is flushed into the systemic circulation, where it functions as a highly active endocrine hormone 3611. Therefore, bone resorption is not merely a catabolic breakdown of skeletal tissue; rather, it serves as an essential endocrine switch required for the systemic release of the active osteocalcin hormone. Conditions or pharmacological interventions that halt bone resorption - such as the administration of bisphosphonates or denosumab - can inadvertently suppress the generation and release of biologically active ucOCN, potentially altering systemic metabolism 14.
Systemic Target Tissues and Metabolic Signaling of Osteocalcin
Pancreatic Beta-Cell Proliferation and Insulin Secretion
The most extensively documented and heavily investigated extra-skeletal function of ucOCN is its profound regulation of whole-body energy metabolism 1315. Once in circulation, ucOCN acts directly on the pancreas by binding to GPRC6A, a highly specialized G-protein coupled receptor (GPCR) that belongs to class C, group 6, subtype A 3616.
Upon activation by ucOCN, GPRC6A triggers intracellular signaling cascades that directly modulate pancreatic $\beta$-cell physiology. The hormone acts as a potent mitogen, promoting rapid $\beta$-cell proliferation and significantly increasing overall $\beta$-cell mass 11517. Furthermore, ucOCN robustly stimulates the synthesis and secretion of insulin, particularly in response to a glucose challenge (glucose-stimulated insulin secretion, or GSIS) 31718.
Simultaneously, ucOCN acts on peripheral metabolic tissues to enhance insulin sensitivity. In adipose tissue, ucOCN signaling upregulates the synthesis and systemic secretion of adiponectin, an insulin-sensitizing adipokine 101418. In the liver, the hormone functions to reduce visceral fat accumulation and significantly limits ectopic lipid storage, thereby offering protective mechanisms against non-alcoholic fatty liver disease (NAFLD) and preventing systemic lipotoxicity 136. Furthermore, ucOCN coordinates with the gut-pancreas axis by stimulating the synthesis of glucagon-like peptide-1 (GLP-1) within the small intestines, providing a synergistic incretin effect that further optimizes postprandial glucose disposal 1319.
Skeletal Muscle Dynamics and Exercise Physiology
The interaction between the skeletal system and musculature is governed by deep biochemical crosstalk mediated through soluble bone-derived factors 2. Skeletal muscle tissue exhibits high sensitivity to undercarboxylated osteocalcin. At the cellular level, ucOCN enhances myocyte nutrient utilization by stimulating both glucose and fatty acid uptake, thereby providing the necessary fuel substrates to support endurance and exercise capacity 319.
Mechanistically, in vitro models reveal that ucOCN promotes myogenic differentiation via the GPRC6A-ERK1/2 signaling pathway, while simultaneously stimulating muscle cell proliferation through the PI3K/Akt/p38 MAPK pathways 2. By driving these anabolic pathways, osteocalcin actively preserves muscle mass and strength, mitigating the physiological decline associated with age-related sarcopenia 2.
This bone-muscle relationship operates as a dynamic, feed-forward physiological loop during physical exertion. During acute aerobic or resistance exercise, contracting skeletal muscle fibers release myokines, most notably Interleukin-6 (IL-6). Muscle-derived IL-6 enters the systemic circulation and binds to specific receptors located on osteoblasts. This binding triggers a rapid increase in the synthesis, processing, and secretion of osteocalcin 20. The resulting burst of circulating osteocalcin then feeds back onto the muscle to increase energy availability and drive structural adaptation, demonstrating that the bone-muscle unit operates as a synchronized, mutually dependent endocrine system 2022.
Central Nervous System and the Acute Stress Response
Osteocalcin is capable of crossing the blood-brain barrier, exerting profound effects on neurodevelopment, cognitive function, and stress physiology 136. Within the central nervous system, ucOCN binds to a distinct receptor, GPR158, which is highly expressed in the hippocampus 3. Activation of the GPR158 pathway promotes neurogenesis, stimulates the synthesis of critical monoamine neurotransmitters (including serotonin, dopamine, and noradrenaline), and limits the expression of GABA-synthesizing enzymes, thereby preventing excessive inhibitory signaling that can impair cognitive processing 36. Animal models and observational data in elderly human cohorts indicate that adequate osteocalcin signaling may protect against age-related cognitive decline and neurodegenerative pathologies, such as Alzheimer's disease, by modulating neuronal energy metabolism and reducing chronic neuroinflammation 615.
Recently, osteocalcin has been identified as a critical physiological mediator of the acute stress response (ASR), an evolutionary mechanism required for vertebrate survival 320. Acute stress rapidly triggers the release of uncarboxylated osteocalcin from the skeleton in a manner entirely independent of the traditional hypothalamic-pituitary-adrenal (HPA) axis. This novel skeletal stress response acts to acutely inhibit parasympathetic tone, unleashing the autonomic responses required to mount a rapid "fight or flight" reaction 320. Concurrently, by stimulating insulin release and enhancing peripheral glucose uptake, the rapid surge in ucOCN counteracts the severe hyperglycemia and extreme insulin resistance normally driven by acute stress-induced spikes in cortisol and catecholamines, effectively buffering the metabolic shock of the stressor 20.
Endothelial Function and Cardiovascular Homeostasis
Beyond metabolic and neurological regulation, osteocalcin exerts direct vasoactive and protective effects on the cardiovascular system. In vitro analyses of human endothelial cells demonstrate that osteocalcin possesses potent anti-apoptotic properties 421. The hormone directly stimulates the phosphorylation of endothelial nitric oxide synthase (eNOS) via the Akt signaling pathway, resulting in a significant increase in nitric oxide (NO) production 421. This increased NO bioavailability promotes vasodilation, reduces vascular resistance, and helps maintain overall vascular homeostasis 421.
Furthermore, ucOCN improves intracellular insulin signaling within vascular smooth muscle cells and demonstrates protective mechanisms against endoplasmic reticulum stress 421. These molecular functions align with clinical observations, where reduced circulating levels of ucOCN are strongly and consistently correlated with advanced atherosclerosis, increased vascular calcification, endothelial dysfunction, and an elevated risk of severe cardiovascular events 36.
The Osteocalcin Knockout Controversy and Methodological Reassessments
The establishment of bone as a systemic endocrine organ was driven heavily by the analysis of complex murine genetic models. However, the exact physiological and metabolic roles of osteocalcin have been the subject of an intense, decade-long scientific debate, primarily centered on stark discrepancies across independent laboratories attempting to replicate early knockout findings 51324.
Initial Phenotypic Characterizations of Knockout Models
The foundational evidence defining osteocalcin as a metabolic hormone originated from the Karsenty laboratory at Columbia University in the mid-2000s. Using a targeted knockout model ($OC^{-/-}$ mice) generated on a mixed 129/SV and C57BL/6J genetic background, early reports indicated that mice lacking the osteocalcin genes displayed a catastrophic metabolic failure 51224. These mice exhibited severe systemic hyperglycemia, profound glucose intolerance, severe insulin resistance, significantly reduced $\beta$-cell proliferation, and increased visceral fat pad mass 513.
Furthermore, male $OC^{-/-}$ mice were reported to suffer from pronounced hypogonadism, exhibiting smaller testes, drastically reduced circulating testosterone levels, and reduced muscle mass compared to their wild-type littermates 113. Corresponding gain-of-function models - such as mice lacking the Esp gene (which encodes OST-PTP, a negative regulator that inhibits OCN decarboxylation) - exhibited the exact opposite phenotype: severe hyperinsulinemia, hypoglycemic tendencies, and robust protection against diet-induced obesity 115.
Independent Replication Failures and Genetic Strain Variations
The extraordinary strength of these initial findings triggered widespread global interest, but subsequent independent attempts to replicate the metabolic phenotypes yielded highly discordant results, plunging the field into controversy 24. Two separate prominent research groups - the Warman laboratory at Boston Children's Hospital and the Komori laboratory at Osaka University - generated their own independent osteocalcin-deficient mouse lines to validate the claims 1324.
The Osaka group utilized advanced genomic techniques to delete both the Bglap and Bglap2 genes, creating a complete functional knockout of osteocalcin 13. Crucially, to ensure strict genetic stability, they backcrossed their mice onto a pure C57BL/6N background for more than eight generations, eliminating the confounding variables associated with mixed-strain models 13. Exhaustive phenotypic analysis of these mice from 11 weeks up to 18 months of age, conducted under both normal and high-fat dietary conditions, revealed an absolute absence of observable abnormalities in glucose metabolism 1213. Fasting plasma glucose, HbA1c levels, body weight, visceral fat accumulation, glucose tolerance, and insulin sensitivity were entirely indistinguishable from wild-type control mice 1213. Furthermore, testosterone synthesis, male fertility, and muscle mass remained completely intact 1213.
The Osaka group concluded that osteocalcin's sole essential function was restricted locally to the bone matrix, where it directed the parallel alignment of apatite crystallites to collagen fibrils to maintain structural integrity, actively refuting its role as a systemic hormone 1213. The Warman group similarly reported a complete inability to detect the metabolic and fertility defects previously published, publicly questioning the validity of the original infusion and metabolic assays 24.
Contemporary Reassessments and Resolution of the Phenotype
The profound divergence in the literature raised serious concerns regarding the reproducibility of skeletal endocrine biology 24. However, comprehensive methodological reassessments published in 2025 and 2026 by Tommassini and Dowd have sought to reconcile these discrepancies by re-evaluating the testing parameters 5.
The reassessment highlighted that methodological nuances - specifically related to the age of the animals, subtle variations in background strain, and laboratory environmental challenges - heavily dictate the penetrance of the metabolic phenotype 522. Early failures to replicate the glucose homeostasis defects, including those utilizing CRISPR/Cas9 models, often analyzed mice at 6 months of age or younger 5. However, highly controlled longitudinal analyses of $OC^{-/-}$ mice strictly on a C57BL/6J background confirmed that while younger males display normal glucose tolerance, the metabolic dysregulation induced by osteocalcin deficiency is characterized by a delayed, age-dependent onset 518.
By 9.5 months of age, male $OC^{-/-}$ mice exhibit statistically significant systemic glucose intolerance, severe insulin resistance, diminished glucose-stimulated insulin secretion (GSIS), and a notably higher ratio of fat pad mass to body weight 518. Crucially, the administration of exogenous uncarboxylated osteocalcin to these aged knockouts successfully rescued the glycemic dysregulation, restoring normoglycemia and significantly lowering circulating blood glucose levels 1718.
These updated findings reaffirm the endocrine role of osteocalcin in glucose metabolism. They strongly suggest that the initial replication disparities were likely the result of strain-specific genetic modifiers (the mixed 129/SV background versus pure C57BL/6 lineages), an incomplete recognition of compensatory metabolic pathways present in young animals, and subtle differences in laboratory dietary potassium and vitamin K statuses that alter baseline carboxylation rates 5822. The contemporary scientific consensus now holds that while bone mineral alignment is definitively controlled by cOCN, the endocrine function of ucOCN is an active physiological reality, albeit one heavily modulated by age, genetic background, and cumulative metabolic stress 81822.
| Study Cohort / Laboratory | Genetic Background | Age of Testing | Metabolic Phenotype Observed | Key Conclusions |
|---|---|---|---|---|
| Karsenty Lab (Original) | Mixed 129/SV & C57BL/6J | 1 to 6 months | Severe glucose intolerance, insulin resistance, low testosterone, high visceral fat. | Established OCN as a critical metabolic and reproductive hormone. |
| Komori Lab (Osaka) | Pure C57BL/6N (Backcrossed >8x) | 11 weeks to 18 months | No metabolic abnormalities. Normal glucose, fat mass, and testosterone. | Refuted endocrine role; restricted OCN function to bone mineral alignment. |
| Tommassini & Dowd (Reassessment) | Pure C57BL/6J | 6 months vs. 9.5 months | Normal at 6 mos; Significant glucose intolerance and insulin resistance by 9.5 mos. | Confirmed endocrine role; proved phenotype is highly age-dependent and strain-sensitive. |
Receptor Pharmacology: GPRC6A Evolution and Population Genetics
Molecular Promiscuity and Isoform Diversity of GPRC6A
If osteocalcin regulates human metabolism, its corresponding receptor must be fully functional. GPRC6A is a highly promiscuous, class C GPCR. In addition to binding osteocalcin, the receptor is activated by a diverse array of ligands, including testosterone, basic L-amino acids (such as L-arginine and L-ornithine), and divalent/trivalent cations like calcium and zinc 192627. This multi-ligand specificity makes GPRC6A a master sensor of nutritional and hormonal states, capable of integrating disparate metabolic signals to coordinate the release of insulin, GLP-1, and IL-6 1927.
The receptor exhibits complex structural diversity. The coding region of the longest isoform of the human GPRC6A gene comprises six exons, with a Venus Fly Trap domain spanning exons 1 to 3, and the transmembrane and C-terminal domains located in exon 6 28. Alternative splicing generates multiple isoforms (e.g., isoforms skipping exon 4 or partially including exon 3) that dictate tissue-specific expression profiles across the brain, skeletal muscle, testes, liver, and pancreas, further modulating ligand sensitivity and downstream signaling cascades 2829.
Evolutionary Divergence of the ICL3 Polymorphism
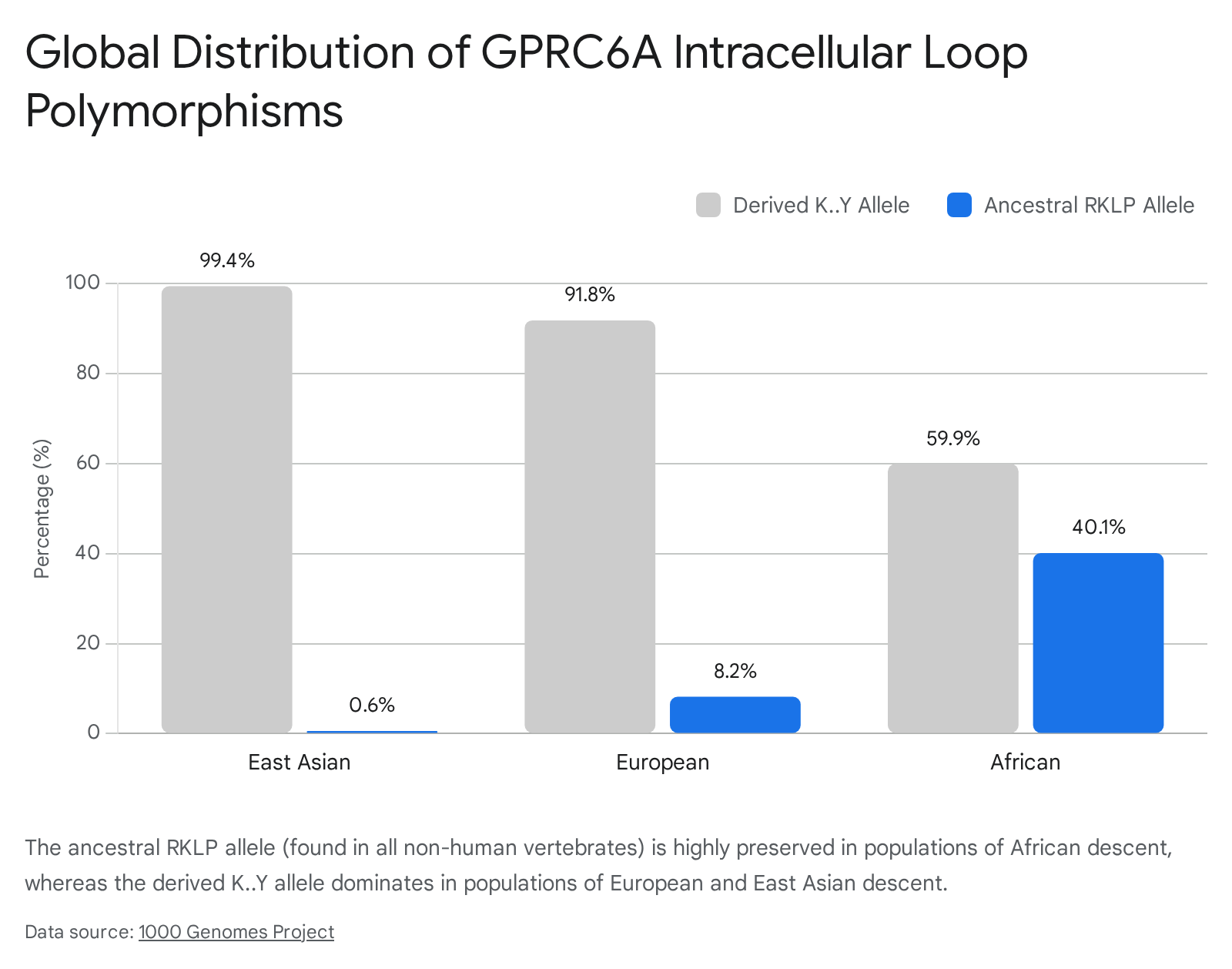
Translating GPRC6A pharmacology from murine models to human physiology has revealed a fascinating and highly controversial evolutionary divergence. Through genomic sequencing databases (such as the 1000 Genomes Project), researchers discovered a widespread, unique polymorphism in the human GPRC6A gene 1923.
In all non-human vertebrate species - including mice, chimpanzees, and the closely related bonobo - the third intracellular loop (ICL3) of the GPRC6A receptor contains a highly conserved amino acid sequence: RKLP (Arg-Lys-Leu-Pro) 192823. However, in modern humans, a dramatic genetic substitution occurred, replacing this ancestral RKLP sequence with a derived "K..Y" sequence (rs386705086) 1928.
The global distribution of this polymorphism highlights distinct population genetics. The uniquely human derived K..Y allele is nearly fixed in populations of East Asian (99.4%) and European (91.8%) descent, as well as in American Hispanic populations (83.4%) 19. Conversely, the ancestral, animal-like RKLP sequence is retained at a much higher frequency (approximately 40%) in populations of African descent 192823.
Debates on Intracellular Retention Versus Gain-of-Function
The physiological consequence of the K..Y substitution has been fiercely debated. Early in vitro studies by the Bräuner-Osborne group suggested that the human K..Y substitution results in a defective receptor that fails to traffic to the cell membrane, becoming permanently retained intracellularly 292331. They argued this evolutionary truncation explains why human clinical trials involving osteocalcin biology often show weak or inconsistent correlations compared to the dramatic phenotypes observed in mouse models 222331.
However, the classification of the human GPRC6A as a non-functional pseudogene has been vigorously contested. Recent, extensive functional assays by the Quarles laboratory demonstrated that human prostate cancer cells and other human tissues naturally expressing the K..Y polymorphic variant successfully localize the receptor to the cell surface membrane 3233. Instead of causing a loss of function, the human K..Y polymorphism fundamentally alters the intracellular signaling bias of the receptor. While the ancestral mouse receptor (RKLP) preferentially signals through the ERK and Akt pathways upon activation by testosterone or osteocalcin, the human K..Y variant acts as a gain-of-function mutation that hyper-activates the mTORC1 pathway 3233.
Implications for Racial Disparities in Metabolic Syndrome
This distinction in receptor signaling carries enormous clinical implications. GPRC6A is highly implicated in prostate cancer tumorigenesis 2632. In vitro and in vivo studies demonstrate that exogenous osteocalcin promotes human prostate cancer proliferation in xenograft models specifically via this hyperactive GPRC6A mTORC1 signaling pathway 32.
Furthermore, the distinct segregation of the ancestral versus derived GPRC6A alleles provides a compelling molecular mechanism to explain established racial disparities in susceptibility to Metabolic Syndrome (MetS) and aggressive prostate cancer 1932. In Indian and South Asian populations, single nucleotide polymorphisms within the GPRC6A gene (such as rs2274911 and F464Y) are highly prevalent and statistically associated with severe metabolic abnormalities and male infertility 223435. Understanding these genetic variations highlights GPRC6A not only as a functional receptor in humans but as a highly viable, population-specific therapeutic target for metabolic and oncological diseases 1932.
Expanded Network of Bone-Derived Hormones
While osteocalcin dominates the literature regarding the bone-pancreas and bone-muscle axes, the skeleton utilizes an entire portfolio of hormones to regulate distinct physiological branches. The endocrine capabilities of bone are further evidenced by Fibroblast Growth Factor 23 (FGF23) and Lipocalin-2 (LCN2) 2424.
Fibroblast Growth Factor 23 and Renal Phosphate Regulation
FGF23 is a 32 kDa hormone produced almost exclusively by osteocytes, the deeply embedded, highly networked mechanosensing cells of the bone matrix 1225. Unlike osteocalcin, which primarily targets glucose and energy metabolism, FGF23 is the master regulator of systemic phosphate homeostasis 125. Under physiological conditions, an increase in dietary or serum phosphate triggers osteocytes to synthesize and release FGF23 into the bloodstream.
The hormone travels to the kidneys, where it binds to FGFR receptor complexes - a process that obligatorily requires the presence of the anti-aging co-receptor Klotho 225. In the renal proximal tubules, FGF23 signaling suppresses the expression of specific sodium-phosphate cotransporters, thereby driving the rapid excretion of phosphate into the urine. Simultaneously, FGF23 inhibits the synthesis of active Vitamin D (calcitriol) to reduce intestinal phosphate absorption 125.
The pathophysiology of FGF23 is highly prominent in Chronic Kidney Disease (CKD) 325. As renal function declines and local Klotho expression drops, the body mounts massive compensatory elevations in circulating FGF23 (often reaching thousands of times above normal levels) to force phosphate excretion through the failing kidneys 25. However, these highly elevated FGF23 concentrations induce severe "off-target" effects. Operating independently of the Klotho co-receptor, toxic levels of FGF23 cause profound left ventricular hypertrophy, cardiac tissue fibrosis, arterial stiffness, and a significantly increased risk of cardiovascular mortality 225.
Lipocalin-2 in Hypothalamic Appetite Suppression
Lipocalin-2 (LCN2) was originally identified and categorized as an adipokine linked to innate immunity and obesity 3839. However, deep molecular tracing has revealed that osteoblasts express and secrete LCN2 at levels at least ten-fold higher than white adipose tissue under basal physiological states 3840.
Osteoblast-derived LCN2 functions as a highly potent anorexigenic (appetite-suppressing) hormone 3839. Upon secretion from bone, LCN2 rapidly crosses the blood-brain barrier, specifically binding to the melanocortin 4 receptor (MC4R) located in the paraventricular and ventromedial neurons of the hypothalamus 383941. Activation of the MC4R pathway strongly inhibits food intake, decreases overall fat mass, and mitigates diet-induced metabolic dysfunction 3840. Consequently, the targeted genetic deletion of LCN2 exclusively in osteoblasts results in severe hyperphagia, profound obesity, systemic glucose intolerance, and pancreatic $\beta$-cell dysfunction in mice 3840.
Autocrine Functions of Lipocalin-2 in Osteocyte Ferroptosis
Emerging evidence highlights a critical intracrine and autocrine role for LCN2 within the bone microenvironment itself. In conditions of systemic obesity, which are frequently accompanied by iron overload, LCN2 acts through its specific receptor, SLC22A17, to tightly sequester intracellular labile iron within osteocytes 42. By regulating these free iron levels, LCN2 prevents excessive lipid peroxidation, oxidative stress, and reactive oxygen species generation 42. This action mitigates iron-mediated cytotoxicity and actively prevents osteocyte ferroptosis (iron-dependent programmed cell death) 42. This unique dual role establishes LCN2 at the crucial intersection of nutrient intake, iron homeostasis, and structural skeletal integrity.
| Bone-Derived Factor | Primary Cellular Origin | Primary Target Organs | Receptors / Pathways | Dominant Systemic Function |
|---|---|---|---|---|
| Osteocalcin (ucOCN) | Osteoblasts | Pancreas, Muscle, Brain, Testes | GPRC6A, GPR158 | Stimulates insulin secretion, muscle glucose uptake; regulates stress and cognition. |
| Fibroblast Growth Factor 23 (FGF23) | Osteocytes | Kidney, Cardiovascular System | FGFR1-4, Klotho co-receptor | Induces renal phosphate excretion; inhibits Vitamin D synthesis; drives cardiac hypertrophy in CKD. |
| Lipocalin-2 (LCN2) | Osteoblasts (also Adipocytes) | Hypothalamus, Bone (autocrine) | MC4R, SLC22A17 | Suppresses appetite via the CNS; regulates intracellular iron to prevent osteocyte ferroptosis. |
| Insulin-like Growth Factor 1 (IGF-1) | Bone Matrix (embedded) | Skeletal Muscle | IGF-1R | Promotes myogenic differentiation and increases muscle mass. |
| Sclerostin (Sost) | Osteocytes | Skeletal Muscle, Bone | Wnt / $\beta$-catenin pathway | Negatively regulates muscle mass and inhibits bone formation. |
Clinical Epidemiology and Global Health Implications
Observational Cohort Associations in Metabolic Disease
The transition from murine genetics and receptor pharmacology to human epidemiology has required the meticulous analysis of vast, diverse population cohorts. Human observational data largely corroborates the existence of the bone-metabolism axis, although the clinical effect sizes are frequently obscured by the natural genetic heterogeneity of human populations, varying analytical methodologies (such as the inability of older assays to differentiate between total OCN and the active ucOCN fraction), and confounding nutritional factors 326.
Cross-sectional and longitudinal cohort studies repeatedly demonstrate that circulating serum osteocalcin levels - specifically the undercarboxylated fraction - are inversely associated with markers of dysmetabolic phenotypes 152745. Lower levels of ucOCN are robustly and independently associated with higher fasting plasma glucose, elevated HbA1c, increased HOMA-IR (indicating severe insulin resistance), higher visceral fat area, hypertriglyceridemia, and a heightened lifetime risk for the development of Type 2 Diabetes Mellitus 6152745. In prospective analyses of elderly adults, exposure to higher osteocalcin levels during follow-up was associated with a significantly lower progressive rise in fasting plasma glucose over a three-year period 27.
Conversely, higher ucOCN concentrations correlate positively with lean body mass, muscular strength (specifically grip and hip flexor strength in elderly cohorts), and superior executive cognitive performance globally 15.
Regional Disparities: Malnutrition, Obesity, and Sarcopenia
These metabolic associations are particularly salient in geographical regions undergoing rapid epidemiological transitions, which currently exhibit a high double-burden of malnutrition and cardiometabolic disease. Meta-analyses mapping the prevalence of Metabolic Syndrome across the African continent reveal an alarmingly high pooled prevalence of 32.4%, with the highest regional rates observed in Southern Africa (33.6%) 4628. This surge is driven by rapidly rising obesity rates and sedentary lifestyles that outpace healthcare infrastructure 4628.
Correspondingly, large demographic sweeps in South Asia and Southeast Asia show an extraordinary prevalence of concurrent Vitamin D insufficiency (exceeding 70% in some Indian and Pakistani cohorts) and early-onset osteopenia, which severely disrupts the normal physiological release of biologically active osteokines 929. In South Asia, the prevalence of osteoporosis ranges from 30% to over 50% among postmenopausal women 929.
In older adult populations spanning from rural Gambia to urbanizing sub-Saharan hubs, high rates of concomitant sarcopenia (affecting up to 30% of rural men) and low bone mass correlate tightly with overall metabolic dysregulation 2930. This synchronized deterioration of bone density and muscle mass reflects a systemic breakdown of the healthy bone-muscle endocrine unit, validating the physiological necessity of osteokine crosstalk in maintaining physical resilience 230.
Therapeutic Avenues: Precision Nutrition and Pharmacological Targeting
The clinical recognition of the skeleton as a highly active endocrine network offers novel therapeutic strategies for managing the twin global epidemics of age-related skeletal fragility (osteoporosis) and metabolic disease (diabetes and sarcopenia) 214.
Nutritional interventions aimed at modulating osteocalcin bioactivity are currently under intense clinical investigation. For instance, chronic supplementation with Vitamin K2 (specifically the MK-7 variant) at dosages ranging from 180 to 375 $\mu$g/day effectively drives the $\gamma$-carboxylation of osteocalcin 3818. While restoring the carboxylated pool (cOCN) is absolutely vital for ensuring proper hydroxyapatite alignment and the prevention of structural fractures, maintaining a delicate physiological balance is required. Excessive over-carboxylation via high-dose supplementation can drastically reduce the circulating pool of the metabolically active ucOCN, potentially blunting its beneficial effects on insulin sensitivity 3818. This highlights the critical need for precision nutrition protocols in clinical skeletal management 818.
Furthermore, recognizing the GPRC6A receptor as a master metabolic regulator provides a clear, highly specific pharmacological target. Designing small-molecule agonists for GPRC6A could theoretically replicate the insulin-sensitizing, muscle-building, and neuroprotective effects of ucOCN 1926. Such targeted medications would be uniquely positioned to bypass the necessity of the skeletal resorption cycle altogether, directly treating the underlying dysfunctions of Metabolic Syndrome by simultaneously targeting $\beta$-cell proliferation, peripheral fat utilization, and the preservation of muscle mass 3619. Ultimately, the ongoing synthesis of osteokine biology reframes bone not merely as an inert victim of aging and metabolic decay, but as a central, highly active regulator whose physiological preservation is essential for long-term whole-body health 2320.