DNA repair capacity theory of aging and its link to lifespan
Principles of the DNA repair capacity theory
The DNA repair capacity theory of aging posits that the progressive deterioration of cellular function, tissue homeostasis, and organismal viability is fundamentally driven by the accumulation of unrepaired damage to the nuclear and mitochondrial genomes. Because DNA serves as the ultimate template for all cellular architecture and metabolic function, maintaining genomic fidelity is critical to survival. The theory dictates that the evolutionary determination of a species' maximum lifespan is tightly correlated with the efficiency, accuracy, and robustness of its inherent DNA repair mechanisms 12.
Every day, the genome of a typical mammalian cell sustains tens of thousands of lesions 3. These arise from endogenous sources, such as reactive oxygen species (ROS) generated by mitochondrial oxidative phosphorylation, spontaneous hydrolytic decay, and DNA replication errors. Exogenous genotoxic agents, including ultraviolet (UV) radiation, ionizing radiation, and environmental chemicals, further compound this burden 34. The resulting lesions span a broad spectrum of structural anomalies: oxidized or alkylated bases, abasic sites, bulky chemical adducts, interstrand and intrastrand crosslinks, single-strand breaks (SSBs), and highly cytotoxic double-strand breaks (DSBs) 35.
To counteract this constant degradation, organisms have evolved an overlapping network of DNA damage response (DDR) pathways. When the capacity of these networks is overwhelmed, macromolecular decay outpaces restorative mechanisms, establishing the biological foundation of aging 12.
Base excision and nucleotide excision repair
The base excision repair (BER) pathway resolves small, non-helix-distorting base lesions, primarily those caused by oxidation, which are particularly prevalent in metabolically active tissues 56. The nucleotide excision repair (NER) pathway is responsible for removing bulky, helix-distorting lesions, such as UV-induced cyclobutane pyrimidine dimers. NER is functionally subdivided into two branches: global genomic NER (GG-NER), which surveys the entire genome for structural distortions, and transcription-coupled NER (TC-NER), which specifically repairs lesions that physically stall RNA polymerase during active gene transcription 478.
Double-strand break repair mechanisms
Double-strand breaks represent the most lethal form of DNA damage. These are resolved via two primary mechanisms: non-homologous end joining (NHEJ) and homologous recombination (HR). HR utilizes a sister chromatid as a template to execute high-fidelity repair, rendering it largely restricted to the S and G2 phases of the cell cycle 89. NHEJ, which simply ligates broken ends together, operates throughout the cell cycle but is inherently error-prone, often introducing small insertions or deletions (indels) at the repair site.
Research evaluating the efficiency of these pathways in mammalian models indicates an age-related decline in overall repair fidelity. While young organisms efficiently utilize standard NHEJ, aging tissues exhibit a progressive reliance on microhomology-mediated end joining (MMEJ), a highly error-prone backup pathway. MMEJ repairs breaks by aligning short, overlapping homologous sequences near the break site, inevitably leading to significant loss of intervening DNA segments and the generation of deleterious chromosomal rearrangements 10.
Evolution of the comparative repair hypothesis
The empirical foundation for the DNA repair capacity theory was established by Hart and Setlow in 1974. By exposing cultured fibroblasts from various mammalian species to UV radiation and measuring unscheduled DNA synthesis - a proxy for excision repair activity - they demonstrated a strong logarithmic correlation between species-specific lifespan and DNA repair capacity 51112.
Subsequent researchers argued that UV-induced repair in cultured fibroblasts is heavily influenced by species-specific ecological factors, such as fur coverage and nocturnal habits, rather than universally applicable aging principles 12. However, the core comparative hypothesis has been robustly validated by modern high-throughput genomics. Transcriptomic analyses of liver tissues - an organ characterized by high oxidative metabolism and abundant spontaneous DNA damage - from mice, naked mole-rats, and humans reveal that long-lived species exhibit significantly higher basal expression of core DNA repair genes, including tumor suppressor TP53, mismatch repair protein MSH3, and NHEJ repair proteins like XRCC6 (Ku70) 12.
Intersection with somatic mutation and information theories
The biological consequences of DNA damage are not limited to direct mutagenesis. The somatic mutation theory of aging, which asserts that aging is driven by irreversible sequence alterations leading to cellular dysfunction and oncogenesis, has been increasingly integrated with epigenetic models of aging 131315. This theoretical synthesis addresses a historical limitation of the somatic mutation theory: the observed mutation burden in aged tissues is often mathematically insufficient to entirely account for the systemic physiological collapse observed in aging organisms 1316.
Epigenetic drift and transcriptional noise
The Information Theory of Aging argues that the primary driver of senescence is the loss of youthful epigenetic information - specifically, the precise patterns of DNA methylation and histone modifications that dictate cellular identity 1514. Unrepaired or continually occurring DNA damage, particularly double-strand breaks, serves as a primary catalyst for this epigenetic drift. When a DSB occurs, chromatin remodeling is required to allow repair enzymes to access the lesion. Epigenetic regulators, including the NAD+-dependent deacetylases SIRT1 and SIRT6, physically migrate from their normal genomic loci (where they maintain gene silencing) to the site of the break to facilitate repair 1415.
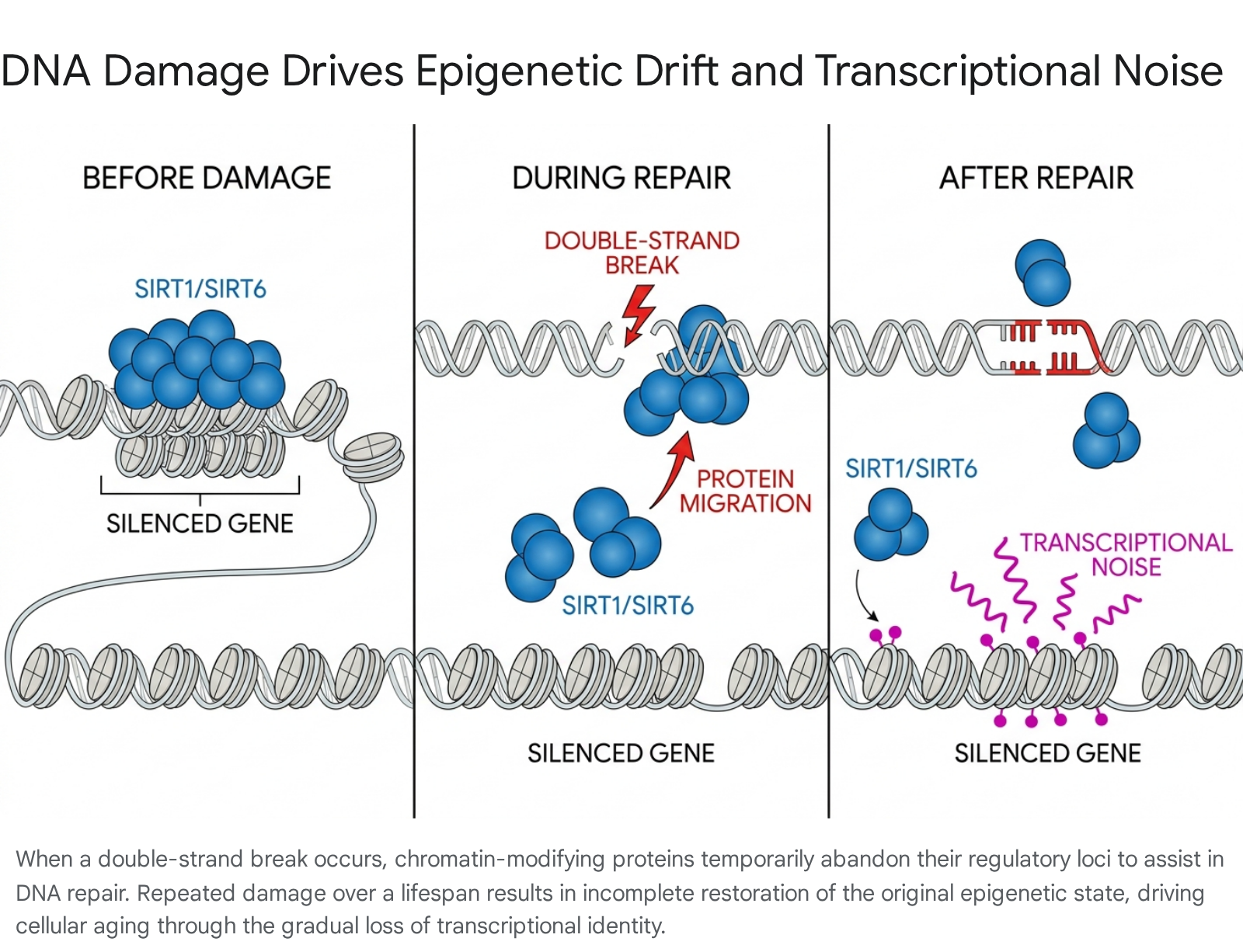
While this mechanism preserves the DNA sequence, it temporarily disrupts the epigenome. In young cells, the epigenetic marks are accurately restored once the repair is complete. However, as the cumulative burden of DNA damage increases over a lifetime, this relocalization process becomes inefficient. Epigenetic modifiers fail to return precisely to their original loci, leading to a permanent redistribution of heterochromatin factors 1416.
This redistribution results in transcriptional noise, characterized by the aberrant expression of normally silenced genes - such as ancient retrotransposons - and the eventual dilution of cellular identity. Under this paradigm, robust DNA repair capacity extends lifespan not merely by preventing oncogenic sequence mutations, but by minimizing the frequency, duration, and severity of epigenetic disruption 151614.
Cellular responses to unrepaired DNA damage
When the burden of DNA damage exceeds the cell's repair capacity, the DDR initiates highly conserved stress signaling cascades. Depending on the cell type, the extent of the damage, and the organism's evolutionary strategy, the cell fate is directed toward transient cell cycle arrest, permanent cellular senescence, or programmed cell death (apoptosis) 1217.
Senescence and the SASP
In highly proliferative mitotic cell populations, such as epithelial and stromal compartments, genomic instability poses a severe oncogenic risk. Unrepaired DSBs or bulky adducts persistently activate the p53 and p21 kinase cascades, forcing the cell into senescence - a state of irreversible growth arrest 218. While senescence acts as a potent tumor-suppressive mechanism in early life, the chronic accumulation of senescent cells (SnCs) becomes a primary driver of tissue dysfunction in late life. Senescent cells secrete a complex, toxic mixture of pro-inflammatory cytokines, chemokines, and matrix metalloproteinases known as the senescence-associated secretory phenotype (SASP) 219.
The cGAS-STING axis
The SASP is deeply intertwined with DNA damage through the cGAS-STING signaling pathway. Cyclic GMP-AMP synthase (cGAS) is a cytosolic DNA sensor primarily evolved to detect viral pathogens. However, severe nuclear DNA damage can result in the leakage of genomic DNA fragments into the cytosol or the formation of micronuclei resulting from unresolved chromosomal aberrations 1620.
cGAS detects this misplaced self-DNA and triggers the STING (stimulator of interferon genes) pathway, igniting a sterile inflammatory response. This overactive immune sensor degrades local tissue architecture, impairs stem cell niches, and accelerates systemic aging. Reducing the activity of this false immune alarm has been shown to restore tissue function in rapid-aging models, directly linking unrepaired DNA to systemic inflammatory aging 161920.
Evidence from human progeroid syndromes
The most compelling human evidence supporting the DNA repair capacity theory stems from rare monogenic disorders collectively termed progeroid syndromes. These conditions are characterized by the accelerated onset of age-related phenotypes - such as alopecia, atherosclerosis, severe osteoporosis, neurodegeneration, and massively increased cancer risk - appearing in childhood or early adolescence 721. The vast majority of these segmental aging syndromes are caused by homozygous or compound heterozygous mutations in genes encoding essential DNA repair enzymes or nuclear envelope proteins necessary for genome maintenance 8.
Nucleotide excision repair defects
The clinical presentation of a progeroid syndrome heavily depends on the specific DNA repair pathway that is compromised. Defects in pathways managing transcription-blocking lesions generally result in severe developmental and neurological degeneration. For instance, approximately 90% of Cockayne Syndrome (CS) cases are caused by mutations in the ERCC8 or ERCC6 genes, which encode the CSA and CSB proteins required for transcription-coupled nucleotide excision repair (TC-NER) 7822.
Because post-mitotic neurons are highly dependent on transcription-coupled repair to maintain active gene expression without replicating, CS patients suffer from profound neurodevelopmental delay, progressive neurological degeneration, and cachexia 422. The Type II form of CS is particularly severe, with an average lifespan of only 5 to 7 years. Conversely, CS patients do not exhibit an increased cancer risk, indicating that other global repair pathways sufficient for tumor suppression remain intact, despite the rapid aging of post-mitotic tissues 48.
A related disorder, Xeroderma Pigmentosum (XP), arises from mutations in genes XPA through XPG, which cripple both global genomic and transcription-coupled NER. XP patients exhibit a 10,000-fold increased risk of non-melanoma skin cancers upon UV exposure. While historical lifespans were extremely short, modern strict UV avoidance can allow XP patients to achieve a normal lifespan, highlighting the environmental dependency of certain DNA damage drivers 782223.
Helicase and double-strand break repair defects
Defects in double-strand break repair and genome replication generally result in high cancer predisposition alongside premature aging. Werner Syndrome (WS) and Bloom Syndrome (BS) are caused by mutations in the RecQ family of DNA helicases (WRN and BLM, respectively), which are critical for preserving genome stability during DNA replication and telomere maintenance 82425.
Werner syndrome features a loss of coordination in BER and HR, leading to rapid telomere attrition, genomic instability, and a dramatic increase in mesenchymal cancers 824. Bloom syndrome, characterized by excessive homologous recombination that destabilizes the genome, drives the development of diverse cancers and reduces the median lifespan to under 30 years 2426.
Nuclear envelope instability
Hutchinson-Gilford Progeria Syndrome (HGPS) offers crucial insight into the spatial and structural organization of DNA repair. HGPS is primarily caused by a de novo single-base substitution in the LMNA gene, generating a toxic, permanently farnesylated protein called progerin 821.
Although progerin is a structural protein rather than a repair enzyme, it severely distorts nuclear architecture. This deformation disrupts peripheral heterochromatin and physically impedes the recruitment of DNA damage response factors, such as RAD51 and 53BP1, to sites of DNA breakage 821. The resulting accumulation of double-strand breaks mirrors the pathology of primary DNA repair defects. HGPS patients typically succumb to myocardial infarction by an average age of 14, reinforcing the principle that functional genome maintenance requires both chemical repair enzymes and a stable topological environment 8.
Summary of progeroid repair deficiencies
The highly specific nature of these genetic diseases demonstrates that the failure of DNA repair pathways recapitulates the exact physiological decline observed in natural aging.
| Syndrome Name | Affected Gene / Protein | Primary Affected Mechanism | Clinical Lifespan Impact |
|---|---|---|---|
| Hutchinson-Gilford Progeria Syndrome (HGPS) | LMNA (Progerin) | Nuclear envelope instability; impaired recruitment of RAD51 and 53BP1 for DNA repair. | Mean survival of ~14 years, typically due to severe cardiovascular disease 821. |
| Cockayne Syndrome (Type I, II, III) | ERCC8 (CSA) or ERCC6 (CSB) | Transcription-coupled nucleotide excision repair (TC-NER) of bulky lesions. | Type I: ~16 years. Type II: ~5 years. Type III: ~30 years. Features severe neurodegeneration but no cancer predisposition 7822. |
| Werner Syndrome (WS) | WRN | RecQ helicase involved in base excision repair (BER) and homologous recombination (HR). | Late adolescent onset of aging features; significant predisposition to mesenchymal cancers 8212425. |
| Bloom Syndrome (BS) | BLM | RecQ helicase regulating homologous recombination to prevent excessive crossover. | Median lifespan <30 years. Extremely high susceptibility to diverse cancers 72426. |
| Xeroderma Pigmentosum (XP) | XPA through XPG | Global genomic and transcription-coupled nucleotide excision repair (NER). | Reduced lifespan historically (<10 years) due to severe skin cancers, but normal lifespan achievable with complete UV avoidance 7822. |
Comparative genomics of extreme longevity models
Evolutionary biology provides naturally occurring models of extreme longevity that challenge the theoretical limits of mammalian aging. Comparative genomic analyses of species that vastly outlive their closely related evolutionary cousins consistently reveal massive selective pressure and adaptive restructuring centered on DNA repair networks 3027.
The Naked Mole-Rat and cGAS mutation
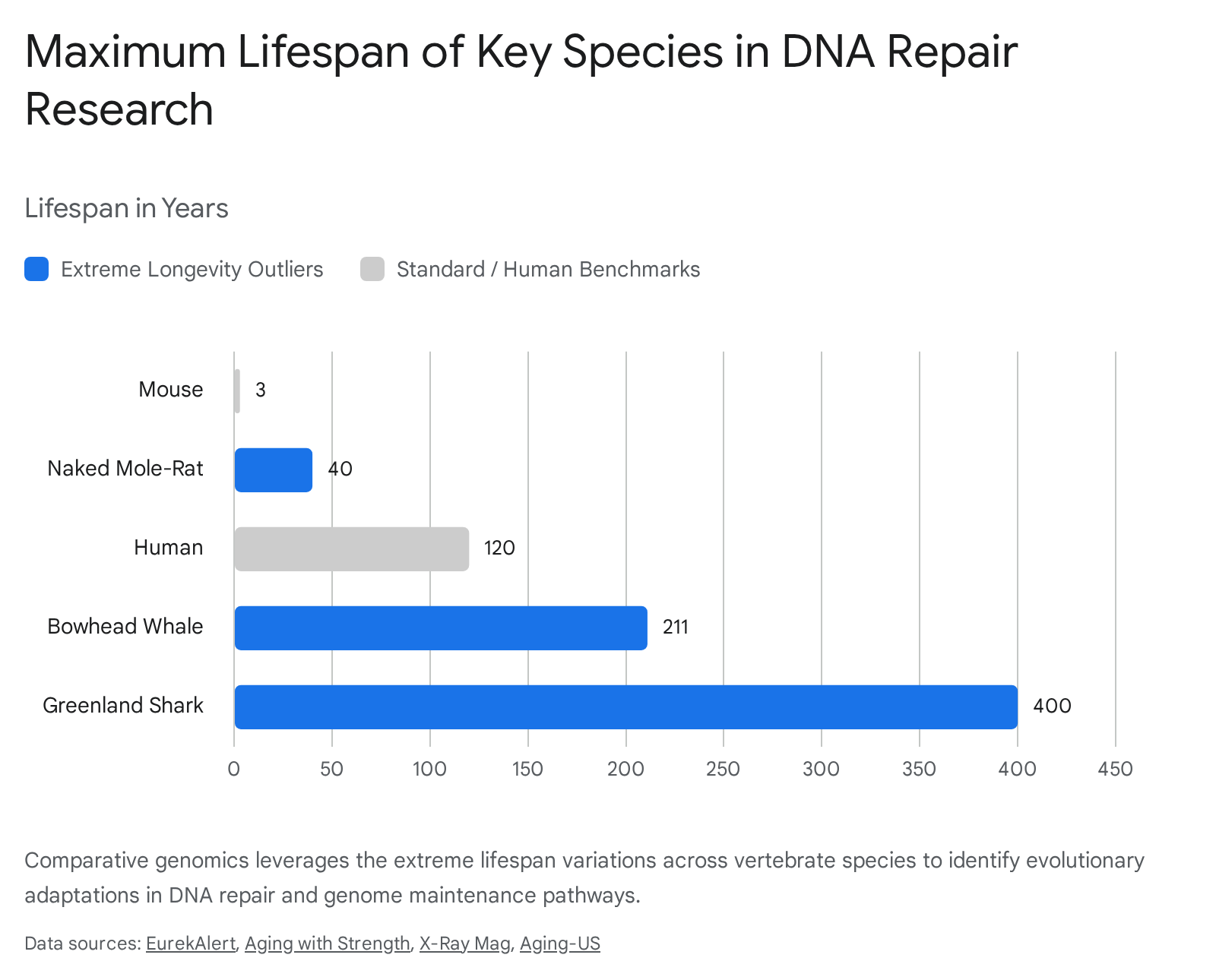
The naked mole-rat (Heterocephalus glaber) is a subterranean rodent with a maximum lifespan approaching 40 years, roughly tenfold that of similarly sized rodents 302829. The species exhibits near-total immunity to cancer and an extraordinary resistance to age-related physiological decline 30.
Recent research has identified unique evolutionary adaptations in the mole-rat's cGAS enzyme. In humans and mice, cGAS suppresses homologous recombination repair when activated, serving as an anti-viral mechanism that prioritizes apoptosis over the repair of a potentially infected cell. In the naked mole-rat, four specific amino acid substitutions in the cGAS protein reduce its ubiquitination and degradation. This alteration allows mole-rat cGAS to persist at higher levels in the nucleus after DNA damage, where it strongly interacts with essential repair factors FANCI and RAD50, thereby boosting the high-fidelity HR repair pathway 2829. Transgenic expression of the mole-rat cGAS variant in fruit flies has been shown to extend lifespan, directly implicating this enhanced DNA repair mechanism in longevity 2829.
The Bowhead Whale and CIRBP expression
Living over 200 years, the bowhead whale (Balaena mysticetus) navigates Peto's Paradox - the observation that cancer incidence does not correlate with body size or lifespan across species, despite massive animals having vastly more cells and cell divisions 30.
Rather than relying solely on enhanced tumor suppression (a strategy utilized by elephants through multiple copies of the TP53 gene), bowhead whales rely heavily on enhanced DNA repair. Genomic analysis reveals that bowhead whale tissues express the CIRBP protein at levels 100 times higher than in other mammals. This protein abundance facilitates dramatically more efficient and accurate repair of DNA damage, actively preventing the accumulation of mutations rather than merely triggering apoptosis in damaged cells 30.
The Greenland Shark and transposable elements
The Greenland shark (Somniosus microcephalus) is the longest-lived vertebrate known, with an estimated maximum lifespan of 400 years 353132. Its 6.5-gigabase genome is exceptionally large, primarily due to a massive expansion of transposable elements ("jumping genes"), which account for over 70% of its genetic code 3233.
While high repeat content usually destabilizes a genome, the Greenland shark has paradoxically leveraged this architecture to expand its DNA repair network. Specifically, 81 genes dedicated to DNA repair have been duplicated across the genome 3133. Furthermore, researchers have identified specific sequence alterations in the shark's TP53 gene, suggesting an enhanced capacity to orchestrate DNA repair and suppress tumors over centuries 3531. Histological examinations of the shark's retinas demonstrate virtually no age-related neurodegeneration even past 130 years of age, highlighting the organism-wide efficacy of this unique repair toolkit 34.
Myotis bats and virus-driven repair adaptations
Bats within the genus Myotis represent another extreme longevity outlier, living decades longer than expected for their metabolic rate and body mass. Genomic sequencing of 8 closely related Myotis species reveals pervasive positive selection in genes related to DNA repair and cancer pathways 4035.
Interestingly, this enhanced DNA damage response appears to be an evolutionary byproduct of immune adaptation. Bats show a genome-wide overrepresentation of positive selection for proteins that interact with DNA viruses. The pleiotropic relationship between adapting to tolerate viral DNA integration and enhancing endogenous DNA repair mechanisms provides a dual benefit. Adaptations in genes like FBXO31 and TP53, alongside trans-species copy number polymorphisms in PKR, confer resistance to infectious zoonotic diseases while simultaneously suppressing the genomic instability that drives aging and cancer 273536.
| Species | Estimated Maximum Lifespan | Primary DNA Repair Adaptation |
|---|---|---|
| Mouse (Mus musculus) | ~3 years | Baseline mammalian repair capacity; prone to rapid telomere attrition and oxidative stress in standard environments 1237. |
| Naked Mole-Rat (H. glaber) | ~40 years | Mutations in cGAS enhance interactions with RAD50/FANCI, boosting high-fidelity homologous recombination 2829. |
| Bowhead Whale (B. mysticetus) | >200 years | 100-fold overexpression of CIRBP protein, drastically increasing the efficiency and accuracy of DNA repair 30. |
| Greenland Shark (S. microcephalus) | ~400 years | Expansion of 81 DNA repair genes driven by transposable elements; highly altered TP53 regulatory networks 353133. |
| Myotis Bats (Myotis spp.) | 30 - 40+ years | Positive selection in cancer/repair pathways driven by an evolutionary arms race with DNA viruses 273536. |
Genomic analysis of human centenarian cohorts
Applying the DNA repair capacity theory to human populations requires investigating individuals who achieve extreme longevity naturally. Studies of centenarians, semi-supercentenarians (105 - 109 years), and supercentenarians (110+ years) reveal an enrichment of rare genetic variants concentrated in genome maintenance pathways 3839.
East Asian longevity cohorts
Extensive research has been conducted on the long-lived populations of Bama County in Guangxi, China, where the proportion of centenarians heavily outpaces international averages. Genetic profiling of the Bama cohort frequently highlights functional polymorphisms in critical DNA repair and metabolic genes, notably SIRT6, hOGG1, XRCC1, and FOXO3A 4041.
The SIRT6 gene is of particular interest due to its multifaceted role in DNA double-strand break repair, chromatin stability, and telomere maintenance. In Bama populations, specific alleles (such as the C allele of the rs350846 polymorphism) are significantly associated with extended lifespan, alongside favorable metabolic profiles including elevated high-density lipoprotein (HDL) and reduced triglycerides 41. Epigenetic profiling of these cohorts also reveals hypermethylation in specific cellular immunity pathways, indicating superior resistance to systemic degradation 40. Analyses of healthy nonagenarians in Azerbaijan similarly underscore the translational resilience of SIRT6; protein-to-mRNA ratios for SIRT6 remain robustly preserved in older age compared to other sirtuins 42.
Furthermore, the transcription factor FOXO3A operates as a master regulator of stress resistance. In long-lived individuals, specific FOXO3A single nucleotide polymorphisms (SNPs) exhibit strong linkage disequilibrium. Advanced chromatin interaction mapping reveals that these variants form long-range physical contacts via chromatin looping (mediated by CTCF binding sites) with 46 neighboring genes over a 7.3 Mb distance on chromosome 6q21. Under genotoxic stress, FOXO3A rapidly physically moves toward these neighboring genes, suggesting that the longevity-associated genotype coordinates a massive, rapid transcriptional response to DNA damage 43.
European cohorts and sex dimorphism
In European cohorts, whole-genome sequencing of 81 Italian semi-supercentenarians and supercentenarians identified a distinct genetic background that renders cellular DNA repair unusually efficient. Compared to younger controls, these extremely long-lived individuals exhibited a significantly lower burden of naturally occurring somatic mutations in key genomic regions 39.
Computational annotations mapped the most frequent genetic changes to the STK17A gene, a kinase involved in coordinating the cellular response to DNA damage, regulating reactive oxygen species, and initiating apoptosis in irreparably damaged cells. The ability of centenarians to tightly manage DNA damage through enhanced STK17A expression is believed to be a central mechanism protecting them from cardiovascular disease, cancer, and cognitive decline 39.
Interestingly, large-scale genome-wide association studies across broad demographics in China and Europe indicate that genetic associations with longevity - including those linked to genome maintenance - are on average statistically stronger in females than in males. Polygenic risk score analyses of sex-specific genes suggest that female genetics derive greater survival benefits from repair variants, partially explaining the universal male-female survival paradox 44.
Tissue-specific variations in DNA repair capacity
While genomic sequencing evaluates an organism's baseline genetic toolkit, the actual execution of DNA repair is highly context-dependent. The physiological consequences of declining repair capacity differ drastically depending on whether a tissue is predominantly mitotic or post-mitotic.
Post-mitotic neurons
Because neurons are generated during early development and must survive for the lifetime of the organism without the ability to self-renew, they represent the ultimate biological test of long-term DNA repair capacity 45. Aging in neurons is characterized by the accumulation of oxidative DNA damage, primarily processed by base excision repair (BER) 64647. Because post-mitotic neurons reside permanently in the G0 phase of the cell cycle, they generally cannot utilize homologous recombination and rely almost entirely on non-homologous end joining (NHEJ) to repair lethal double-strand breaks 647.
Recent applications of a novel sequencing technique, "Repair-seq", have revealed that aged neurons do not repair their DNA randomly. Instead, they exhibit distinct repair "hot spots." Researchers identified approximately 61,000 such regions, comprising just 1.6% of the neuronal genome, which are heavily prioritized by the repair machinery. Crucially, these hot spots contain genes essential to maintaining neuronal identity and function 45. The progressive failure to maintain these specific high-priority regions correlates strongly with the pathological protein changes observed in neurodegenerative conditions like Alzheimer's disease 45.
Furthermore, studies applying CRISPR-Cas9 to induce programmed DNA breaks in human induced pluripotent stem cell (iPSC)-derived neurons have uncovered fundamentally different repair kinetics compared to dividing cells. While stem cells repair DSBs within hours or days, post-mitotic neurons take up to two weeks to resolve identical lesions 4654. During this prolonged repair window, neurons mount an atypical gene expression response, upregulating replication-associated factors like RRM2 (a ribonucleotide reductase subunit) to facilitate repair without formally re-entering mitosis. This slow, deliberate repair timeline makes neurons uniquely vulnerable to chronic genotoxic stress 4654.
Neuronal resilience is also influenced by genetic variants outside canonical repair genes. The APOE2 variant, a known protective factor for exceptional longevity and a buffer against Alzheimer's disease, has recently been shown to play a direct role in nuclear stability. Human neurons expressing APOE2 exhibit strongly upregulated DNA repair pathways, lower baseline DNA damage, and better-preserved heterochromatin compared to neurons expressing the high-risk APOE4 variant, directly linking lipid metabolism genes to genome maintenance in the aging brain 48.
Cardiomyocytes
Like neurons, adult cardiomyocytes are terminally differentiated, post-mitotic cells. The heart is subjected to continuous, life-long mechanical stress due to contractile activity. This biomechanical force is transduced directly to the cardiomyocyte nucleus, rendering the DNA highly vulnerable to physical rupture and mechano-transduced damage, in addition to standard oxidative stress 49.
When DNA damage persists in the heart, it triggers maladaptive cardiac remodeling. Unrepaired DNA damage activates classical senescence pathways (p21 and p16), leading to cardiomyocyte hypertrophy, mitochondrial dysfunction, and fibrosis 4950. Experimental murine models with systemic or cardiomyocyte-specific knockouts of essential NER endonucleases (such as ERCC1 or XPG) demonstrate that while these mice undergo normal embryonic heart development, the inability to repair spontaneous endogenous DNA damage rapidly drives the early onset of severe congestive heart failure and premature death within 6 months 1851.
Translational gaps between in vitro and in vivo models
Much of the foundational knowledge regarding cellular senescence and DNA repair capacity was derived from in vitro studies of human diploid fibroblasts (e.g., WI-38 cells). Historically, the progressive loss of proliferative capacity in culture - the Hayflick limit - was equated directly with organismal aging 3752. However, significant translational gaps exist between these models and physiological aging.
In standard tissue culture, fibroblasts are grown under atmospheric oxygen tension (~20% O2), which is drastically higher than physiological tissue normoxia (~3% O2). This hyperoxic shock induces massive amounts of unphysiological oxidative DNA damage, forcing cells into premature, stress-induced replicative senescence after only a few passages 5354. While fibroblasts extracted from elderly human donors do exhibit impaired redox balances, delayed migration, and distinct translational profiles compared to those from young donors, the rapid cascade of organelle failure seen in late-passage cultured cells is quantitatively and qualitatively distinct from natural, in vivo aging 375355.
Furthermore, advanced ribosome profiling in progeroid (Ercc1Δ/ - ) mouse models has clarified the specific mechanisms of in vivo decline. While the accumulation of DNA damage physically stalls RNA polymerase - thereby skewing the transcriptional landscape in a gene-length-dependent fashion - the translational efficiency of the surviving mRNA remains largely intact 56. This indicates that the ultimate functional failure in aged organisms is driven primarily by the DNA damage-induced loss of accurate transcription, rather than a global breakdown of the protein synthesis machinery itself 1756. Therefore, interventions aiming to delay aging must act upstream at the level of the genome and epigenome.
Emerging therapeutic interventions targeting DNA repair
Recognizing DNA damage and repair failure as root causes of aging has catalyzed the development of geroprotective therapeutics aimed at artificially enhancing genome maintenance 219.
Pharmacological modulation of sirtuins and NAD+ metabolism is currently a primary focus. The efficiency of base excision repair and DSB repair relies heavily on PARP1, an enzyme that senses breaks and consumes massive quantities of NAD+. Supplementation with NAD+ precursors (like nicotinamide riboside) restores PARP1 activity and prevents the metabolic collapse associated with DNA damage. Concurrently, small-molecule activators of SIRT6 (such as UBCS039) are being investigated to enhance the recruitment of DNA repair complexes to double-strand breaks, thereby improving genomic stability and extending healthspan 194257.
Gene regulation therapies are also advancing. The DREAM repressor complex suppresses the expression of numerous DNA repair genes in post-mitotic cells. Recent studies demonstrate that inhibiting the DREAM complex via DYRK1A inhibitors (e.g., harmine) robustly upregulates multiple repair pathways simultaneously - including BER, NER, and HR - supercharging the cell's intrinsic capacity to resolve lesions 19.
Finally, cellular reprogramming offers a method to reverse DNA damage-induced epigenetic noise. Ectopic expression of the Yamanaka factors (Oct4, Sox2, Klf4) in vivo has been shown to induce partial epigenetic reprogramming. By restoring a youthful epigenetic state, this intervention actively promotes more efficient DNA repair and reverses age-related decline in tissues such as the optic nerve and skeletal muscle, directly counteracting the epigenetic degradation generated by a lifetime of DSB repair 151958.
Conclusion
The DNA repair capacity theory of aging has evolved from a simple correlation between excision repair and lifespan into a highly complex, multi-dimensional framework. It is now understood that unrepaired DNA damage drives organismal decline through multiple intersecting vectors. It generates oncogenic mutations, physically impedes the transcription of essential genes in post-mitotic tissues like the brain and heart, triggers sterile systemic inflammation via the cGAS-STING pathway, and permanently disrupts the epigenome by forcing chromatin modifiers to constantly migrate to sites of injury. The natural world provides proof-of-concept that aging can be delayed; extreme longevity models - from the bowhead whale's abundance of CIRBP to the unique viral adaptations of bats - demonstrate that enhancing DNA repair capacity is biology's primary strategy for extending lifespan. As molecular understanding deepens, translating these evolutionary adaptations into pharmacological interventions presents one of the most promising frontiers in modern gerontology.