Clonal hematopoiesis and cardiovascular disease
1. Introduction: The Convergence of Hematology and Cardiovascular Medicine
Historically, the aging process has been associated with an accumulation of traditional cardiovascular risk factors, culminating in atherosclerotic cardiovascular disease (ASCVD), heart failure, and arrhythmias. However, over the past decade, and accelerating rapidly with comprehensive updates in 2024, 2025, and 2026, the discovery of clonal hematopoiesis of indeterminate potential (CHIP) has fundamentally redefined the pathophysiological understanding of aging, hematologic malignancy, and cardiometabolic health 1234.
CHIP is an age-related phenomenon defined by the clonal expansion of hematopoietic stem cells (HSCs) harboring somatic mutations in recognized leukemia driver genes - most commonly DNMT3A, TET2, ASXL1, and JAK2 - in individuals presenting without overt hematologic malignancies or unexplained cytopenias 2456. While originally identified as a precursor state conferring an elevated relative risk for hematologic cancers, subsequent epidemiological and mechanistic analyses have demonstrated that the primary driver of excess mortality in CHIP carriers is not oncological, but rather cardiovascular 7889.
The presence of CHIP independently confers a two- to four-fold elevated risk of incident ASCVD, myocardial infarction (MI), and stroke, rivaling the risk magnitude of established traditional factors such as hypertension, diabetes mellitus, and hyperlipidemia 7810. Recent data published in leading cardiology and hematology journals - culminating in the 2026 American Heart Association (AHA) Scientific Statement on Clonal Hematopoiesis and Cardiovascular Implications - have exponentially expanded the scope of CHIP-related pathologies to include heart failure with preserved ejection fraction (HFpEF), atrial fibrillation (AF), and venous thromboembolism (VTE) 341113.
This report provides an exhaustive, evidence-based analysis of the CHIP landscape. It examines the evolving diagnostic boundaries separating pre-malignant states in current hematopathology frameworks, explores the marked geographic and ancestral disparities in clonal architecture, maps distinct genetic mutations to their specific cardiovascular and inflammatory mechanisms, and evaluates the clinical debate surrounding screening and targeted therapeutics.
2. Taxonomic Evolution: Delineating CHIP, CCUS, and Myeloid Neoplasms
The nosology of age-related clonal hematopoiesis has undergone a significant paradigm shift following the parallel releases of the World Health Organization (WHO) 5th Edition classification (2022, finalized in 2024) and the International Consensus Classification (ICC) of Myeloid Neoplasms and Acute Leukemias (2022, finalized in 2024 and 2025). The precise delineation of these entities is of critical importance to both hematologists and cardiologists, as therapeutic pathways, clinical trial eligibility, and prognostic implications diverge sharply across the spectrum from benign clonal expansion to overt malignancy 5121513.
2.1 Diagnostic Criteria for Clonal Hematopoiesis of Indeterminate Potential (CHIP)
According to the unified frameworks of both the WHO and the ICC, a clinical diagnosis of CHIP requires the detection of one or more somatic mutations in leukemia-associated driver genes with a variant allele frequency (VAF) of ≥2% (or ≥4% for X-linked gene mutations in male patients) in the DNA of peripheral blood or bone marrow cells 51215. Crucially, the individual must exhibit an absolute absence of unexplained persistent cytopenias and an absence of morphological diagnostic criteria for defined myeloid neoplasms 56.
While ultra-deep, high-sensitivity next-generation sequencing approaches can detect minute clones with a VAF <2% (informally termed "micro-CHIP" or generalized age-related clonal hematopoiesis [ARCH]) in nearly all aging adults, the ≥2% threshold remains the accepted clinical and biological standard 2617. Clones exceeding this threshold, and particularly those with a VAF >10%, correlate with significantly elevated cardiovascular and oncologic risks 61714. The detection of CHIP frequently coincides with alterations in DNA methylation patterns, establishing "epigenetic clocks" that estimate epigenetic age acceleration; research indicates that an increase in CHIP clone size correlates directly with increased epigenetic age acceleration, cementing CHIP as a surrogate marker of advanced biological aging 1415.
2.2 Progression to Clonal Cytopenia of Undetermined Significance (CCUS)
Clonal Cytopenia of Undetermined Significance (CCUS) represents a more advanced biological state along the leukemogenic continuum. CCUS is formally diagnosed when a CHIP-qualifying driver mutation (VAF ≥2%) is identified concurrently with one or more persistent, unexplained cytopenias 613. The ICC has established strict cytopenia thresholds that must endure for at least four months to satisfy this diagnosis: hemoglobin concentrations <13 g/dL in males or <12 g/dL in females, absolute neutrophil counts <1.8 * 109/L, and/or platelet counts <150 * 109/L 61513.
Identifying CCUS is of paramount clinical importance, as the simultaneous presence of cytopenia and clonal hematopoiesis dramatically accelerates the risk of progression to myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) compared to CHIP alone 516. The Clonal Hematopoiesis Risk Score (CHRS), established as a prognostic tool, integrates variables such as the presence of specific high-risk mutations (e.g., TP53, U2AF1, SRSF2, SF3B1, IDH1/2), the detection of multiple mutations, clone sizes ≥20% VAF, and elevated mean corpuscular volume (MCV) or red cell distribution width (RDW) to classify individuals into low, intermediate, and high-risk progression groups 5.
2.3 The Boundary of Early Myelodysplastic Syndromes (MDS)
The precise boundary separating CCUS from early MDS has been a major focal point of recent updates in hematopathology, revealing notable divergences between the WHO 5th Edition and the ICC. Sequencing data alone is universally considered insufficient to differentiate CCUS from early-stage MDS; a comprehensive bone marrow morphological assessment via aspirate and biopsy remains mandatory 616.
The WHO 5th Edition strictly requires the presence of morphologic dysplasia - defined as ≥10% dysplastic cells in at least one hematopoietic lineage (myeloid, erythroid, and/or megakaryocytic) - to establish a definitive diagnosis of MDS (now termed myelodysplastic neoplasm, though the abbreviation MDS is retained to avoid confusion with myeloproliferative neoplasms) 1316.
In a significant pathophysiological divergence, the ICC permits a diagnosis of MDS even in the complete absence of morphologic dysplasia if specific, highly penetrant disease-defining genetic abnormalities are present. These include biallelic TP53 mutations, isolated del(5q), or SF3B1 mutations 121317. Consequently, certain patients presenting with cytopenia and a clonal mutation but lacking visible dysplasia might be classified as having CCUS under the WHO framework, but formally diagnosed with MDS under the ICC framework 1317.
Furthermore, the ICC introduced the novel category of "MDS/AML" to classify cases exhibiting 10% to 19% blasts in the peripheral blood or bone marrow, eliminating the former "MDS with excess blasts, grade 2" category to allow these patients access to both advanced MDS and AML clinical trials 1218. The ICC also integrated acute erythroid leukemia (AEL) - previously pure erythroid leukemia - into the broader category of AML with mutated TP53, recognizing the profound biological overlap between these entities 1218. These nuanced taxonomic shifts carry profound therapeutic implications, dictating the transition from watchful waiting in CCUS to the initiation of disease-modifying therapies or allogeneic stem cell transplantation in MDS 16.
3. The Epidemiology of CHIP: Age Stratification and Clone Size Dynamics
CHIP is fundamentally a pathology of the aging hematopoietic system. Driven by the lifelong accumulation of somatic mutations, the competitive advantage of mutant clones, and the shifting selective pressures of the aging bone marrow microenvironment, the prevalence of CHIP exhibits an exponential trajectory across the human lifespan 14.
The prevalence of Clonal Hematopoiesis of Indeterminate Potential (CHIP) rises sharply with advancing age, exhibiting an exponential trajectory after the fifth decade of life and affecting upwards of 30% to 50% of adults over the age of 80 1719.
While cross-sectional analyses of the general population reveal a baseline prevalence that increases steeply with each passing decade, CHIP is notably prevalent even in younger cohorts presenting with premature disease states. For instance, high-sensitivity targeted screening of young adults (aged 18 to 60) presenting with ischemic stroke of undetermined etiology revealed an elevated CHIP prevalence of 20.8%, which was three- to four-fold higher than age-matched controls in the general population 719. This suggests that while CHIP is driven by chronological aging, accelerated biological aging and the presence of subclinical atherosclerosis may bidirectionally facilitate clonal expansion 714.
Table 1: Age-Stratified Prevalence of CHIP (VAF ≥2%) in the General Population
| Age Bracket (Years) | Estimated Prevalence | Key Characteristics & Dominant Mutational Landscape |
|---|---|---|
| 18 - 40 | < 1.0% | Exceedingly rare in the general population; highly associated with premature cardiovascular events, prior cytotoxic therapies, or inherited germline predispositions if detected. |
| 40 - 50 | ~ 5.0% - 6.5% | Early expansion phase; DNMT3A emerges as the most frequently observed and dominant driver mutation. |
| 50 - 60 | 10.0% - 15.0% | Rapid inflection point of incidence; robust epidemiological associations with early-onset coronary artery disease and accelerated atherosclerosis begin to manifest clinically. |
| 60 - 70 | 15.0% - 25.0% | High prevalence; variant allele frequencies (VAF) frequently expand beyond the 10% threshold, correlating with highly significant absolute cardiovascular and cardiometabolic risks. |
| 70 - 80 | 20.0% - 30.0% | Advanced aging phenotype; mutations in splicing factors (SF3B1, SRSF2, U2AF1) and TET2 mutations become highly prevalent alongside established DNMT3A clones. |
| 80+ | 30.0% - 50.0%+ | Near-ubiquitous depending on sequencing sensitivity; the presence of multiple concurrent mutations and mosaic chromosomal alterations (mCAs) are frequent, driving steep increases in all-cause mortality and heart failure. |
Data synthesized from longitudinal population genomic cohorts, the UK Biobank, and contemporary 2023-2026 cardiovascular and hematology literature 1719. Note: Specific prevalence rates are heavily dependent on the depth of coverage and analytical sensitivity of the sequencing platform utilized.
4. Ancestral, Geographic, and Disparate Cohort Landscapes
Historically, massive genomic association studies assessing clonal hematopoiesis have been heavily skewed toward populations of Northern European descent, relying predominantly on databases such as the UK Biobank or Icelandic cohorts 202127. However, recent 2024 and 2025 analyses leveraging highly diverse global biobanks have uncovered profound racial, ethnic, and geographic disparities in both the overall prevalence of CHIP and its underlying mutational architecture 2022.
4.1 African Ancestry and the "All of Us" Research Program
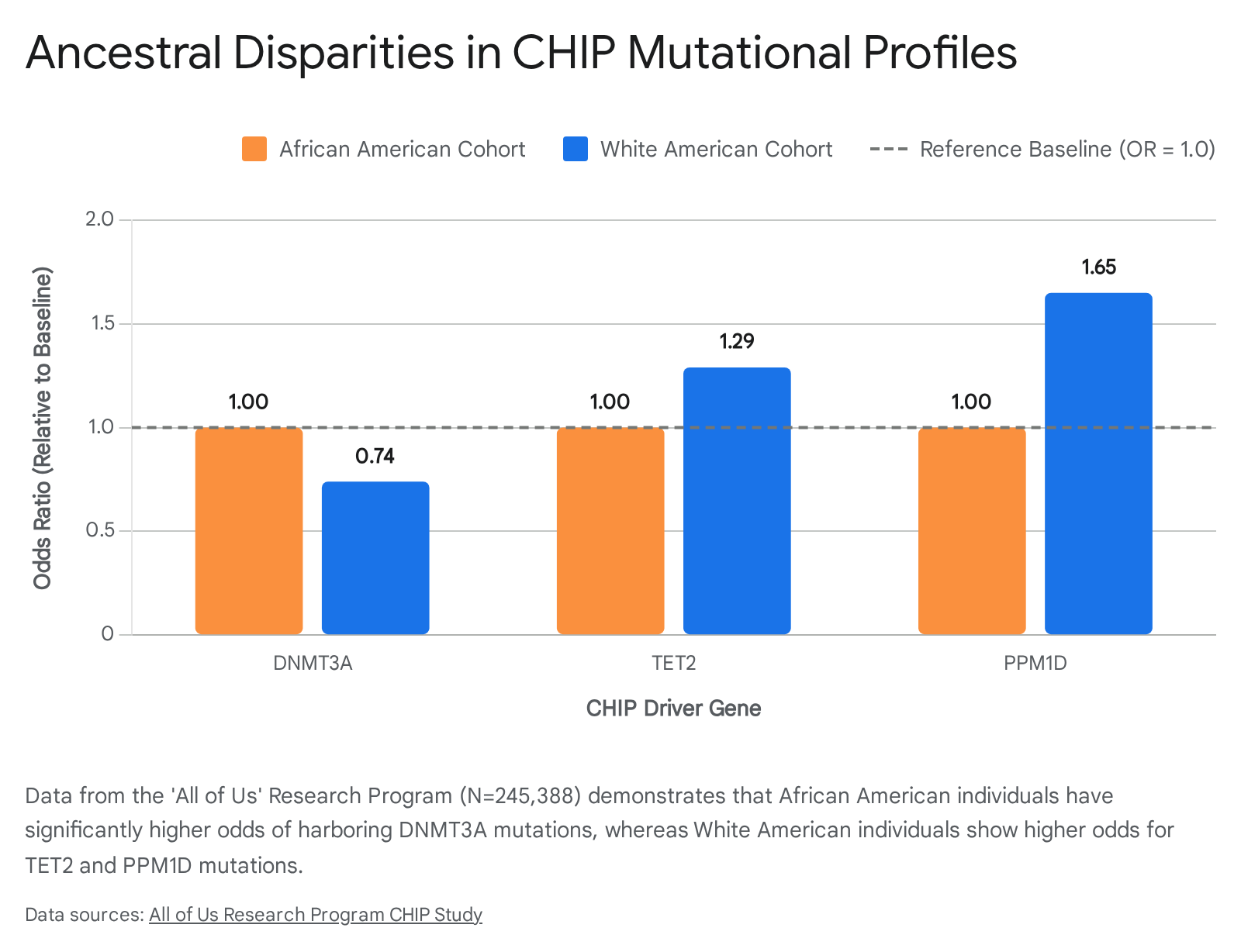
The United States-based "All of Us" Research Program, comprising an analysis of 245,388 racially diverse participants published in late 2025, highlighted stark ancestral divergences. African American (AA) individuals exhibited significantly higher overall odds of harboring CHIP compared to White American (WA) individuals, an association that persisted after rigorous adjustment for age, sex, and smoking status 20.
The genetic drivers of these clonal expansions were highly distinct. White American individuals were significantly less likely to carry DNMT3A mutations (Odds Ratio [OR] = 0.74), specifically the R882H hotspot variant, compared to African American individuals 20. Conversely, WA individuals demonstrated higher odds of harboring TET2 (OR = 1.29) and PPM1D mutations (OR = 1.65) 20.
These mutational disparities translated directly into divergent clinical trajectories. While CHIP predictably increased the risk of hematologic malignancies (HM) across all demographics, the specific phenotypic outcome was heavily influenced by ancestry. In African American individuals, CHIP - particularly when driven by JAK2 mutations - was significantly more strongly linked to the development of myeloproliferative neoplasms (MPNs) (Hazard Ratio [HR] = 3.62) than in White American individuals (HR = 1.84) 20. Furthermore, complementary 2024 exome sequencing data from Black patients with acute myeloid leukemia (AML) revealed a highly distinct mutational landscape, identifying previously unreported PHIP mutations in 7% of patients and establishing differing prognostic indicators for NPM1 and NRAS mutations compared to cohorts of European descent 23.
4.2 South Asian and East Asian Cohorts
Epidemiological investigations into East Asian cohorts highlight powerful intersections between CHIP, progressive cardiovascular phenotypes, and baseline metabolic risk. A robust 2024 study originating from BioBank Japan and related regional registries evaluated 1,004 Japanese patients with atrial fibrillation (AF) against healthy controls. CHIP mutations (VAF ≥2.0%) were detected in 23.6% of AF patients compared to only 10.7% in non-AF subjects 24. TET2 somatic mutations presented the highest specific association with AF (Adjusted OR 1.65), correlating clinically with severe left atrial enlargement, prolonged AF duration, and significant diastolic dysfunction 24.
In a cohort of 4,300 asymptomatic Korean participants, the presence of CHIP demonstrated a striking, mathematically synergistic effect when combined with conventional hyperlipidemia. CHIP carriers with concurrently high LDL cholesterol levels exhibited an over 6-fold amplified risk of ASCVD (HR 6.20), driving the rapid formation of highly unstable, necrotic plaques in proximal coronary lesions during the earliest stages of atherosclerosis 25.
South Asian populations, while historically underrepresented in CHIP registries, face uniquely high burdens of cardiometabolic disease, prompting targeted interventions like the SAHELI (South Asian Healthy Lifestyle Intervention), SAHARA, and U-SACHI (Utah South Asian Cardiovascular Health Initiative) trials 26. Recent multi-ancestry genomic analyses indicate a 1.2% prevalence of pathogenic cardiomyopathy gene variants in individuals of South Asian descent, significantly higher than the 0.7% general population average, underscoring the urgent need to map the specific interplay of CHIP alongside endemic cardiovascular vulnerabilities in these groups 2633.
4.3 Latin American Populations and the GLAD Project
Latin American populations exhibit high degrees of genetic admixture and have remained critically underrepresented in precision medicine and polygenic risk score developments 2727. Recent initiatives, most notably the Genetics of Latin American Diversity (GLAD) Project - which compiled genome-wide information from 53,738 Latin Americans across 39 distinct studies in 2024 - aim to rectify this disparity 27. Initial meta-analyses assessing CHIP across diverse demographics suggest that individuals of Hispanic or Latino descent generally exhibit a lower overall risk of harboring CHIP compared to Non-Hispanic White or Black populations 222829. The exact mechanisms driving this relative biological protection remain poorly understood, though hypotheses center on complex admixtures of genetic resilience, differing environmental and lifestyle exposure timelines, and variations in historical cytotoxic stressors 2728.
5. Molecular Pathways: Mapping Gene Mutations to Cardiovascular Pathophysiology
The epidemiological link between age-related clonal hematopoiesis and cardiovascular disease is not a uniform biological phenomenon. The magnitude of the cardiovascular risk, the specific disease phenotype, and the underlying cellular pathways are highly dependent on the precise mutant gene driving the expansion of the hematopoietic clone 110. The overwhelming majority of CHIP cases are driven by mutations in epigenetic regulators (DNMT3A, TET2, ASXL1), while a highly potent, albeit smaller, subset is driven by signaling kinases (JAK2) or DNA damage response genes (TP53, PPM1D) 830.
Table 2: Comparative Mapping of Major CHIP Driver Genes to Cardiovascular Mechanisms
| Driver Gene | Normal Cellular Function | Pathophysiological Mechanism in CHIP | Primary Cardiovascular Phenotype |
|---|---|---|---|
| TET2 | DNA demethylation (critical epigenetic regulation and gene expression). | Loss of function prevents DNA demethylation, leading to the transcriptional derepression of key inflammatory genes. Triggers massive, aberrant activation of the NLRP3 inflammasome, resulting in the overproduction of IL-1β and IL-6 by mutant macrophages in the arterial wall. | Accelerated atherosclerosis, highly vulnerable necrotic lipid plaques, incident HFpEF, and atrial fibrillation 103132. |
| DNMT3A | DNA methylation (epigenetic silencing of developmental and inflammatory loci). | Loss of function disrupts normal chromatin silencing. Drives a robust pro-inflammatory milieu via enhanced expression of IL-6, CXCL1, and CXCL2 in circulating monocytes and T cells. Increases macrophage infiltration into the vascular wall. | Incident coronary artery disease, early-onset myocardial infarction, general heart failure progression 10. |
| ASXL1 | Chromatin remodeling (interacts heavily with PRC1/PRC2 complexes and EZH2). | Loss of function disrupts H3K27me3 repressor marks. Skews macrophage differentiation and alters DNA methylation landscapes. Promotes systemic inflammation via the EGR1-TNF-α axis. Alters mitochondrial metabolism and drives pathogenic proteomic signatures. | Advanced atherosclerotic risk, highly reduced left ventricular ejection fraction (HFrEF), widespread vascular damage, poor overall survival 9333435. |
| JAK2 (V617F) | Tyrosine kinase signaling (fundamental control of hematopoiesis and cell division). | Gain of function causes cytokine-independent myeloproliferation. Generates hyperreactive, reticulated young platelets. Upregulates COX-1/2 and Thromboxane A2, inducing cross-talk that directly activates adjacent normal wild-type platelets. Primes circulating neutrophils for NETosis. | Severe arterial thrombosis (MI, ischemic stroke) and profound venous thromboembolism (VTE/PE). Confers the highest per-mutation absolute ASCVD risk 103637. |
5.1 The Inflammasome Axis: TET2 and DNMT3A
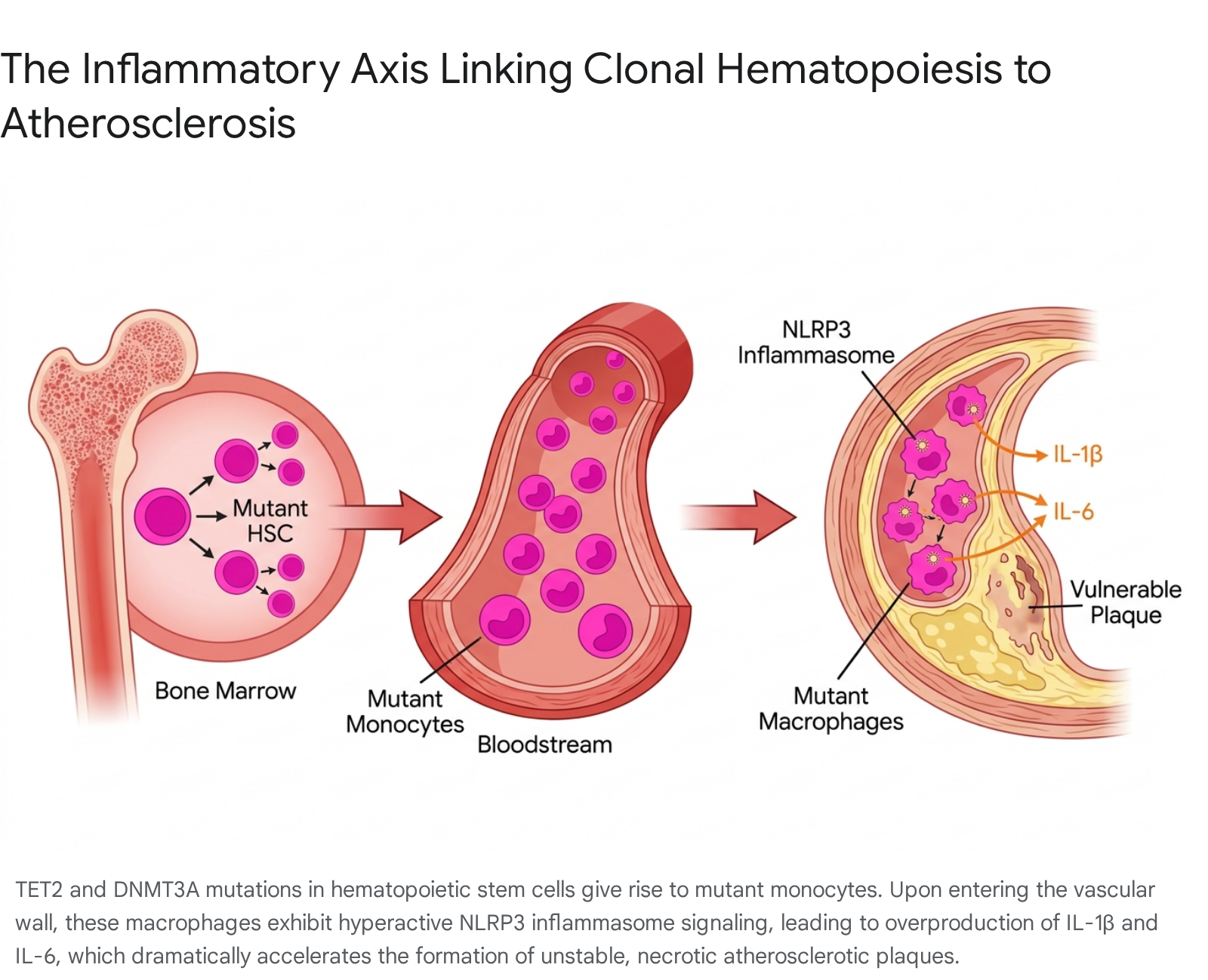
TET2 and DNMT3A dominate the epidemiological landscape as the most frequently mutated genes in CHIP 91430. Both function primarily as epigenetic modifiers; DNMT3A orchestrates de novo DNA methylation to silence genes, while TET2 catalyzes the oxidation of methylcytosine to promote DNA demethylation 810. Their loss-of-function alters the foundational methylation landscape of hematopoietic stem cells, passing profound epigenetic scars down to terminally differentiated progeny - most notably circulating monocytes and tissue-resident macrophages 810.
When these epigenetically deregulated mutant macrophages migrate into the vascular intima and encounter local stressors, such as oxidized low-density lipoproteins or cholesterol crystals, they exhibit a hyper-inflammatory response. In TET2-deficient cells, the lack of epigenetic repression leads directly to the pathological activation of the NOD-like receptor protein 3 (NLRP3) inflammasome 1045. This massive multiprotein complex cleaves pro-interleukin-1β (IL-1β) into its active form, which is rapidly secreted and subsequently stimulates the massive downstream systemic production of interleukin-6 (IL-6) 104546. This specific IL-1β/IL-6 inflammatory axis acts locally to accelerate atherogenesis, promotes the formation of exceptionally large, necrotic lipid cores within the vascular endothelium, and drives chronic, systemic endothelial dysfunction 810.
5.2 Chromatin Remodeling and Systemic Disruption: ASXL1
ASXL1 mutations represent a particularly aggressive and deleterious CHIP subtype, frequently co-occurring with adverse clinical outcomes across both hematology and cardiology 935. While functioning as an epigenetic regulator, ASXL1 operates distinctly from TET2 and DNMT3A by interacting intricately with the Polycomb Repressive Complex 2 (PRC2) - specifically its core component EZH2 - to mediate H3K27me3, a crucial repressive histone modification associated with gene silencing 35.
The loss of ASXL1 expression results in profound transcriptomic dysregulation that extends far beyond the traditional IL-1β/IL-6 pathway. Recent 2025 studies have demonstrated that ASXL1-driven CHIP is uniquely characterized by highly elevated high-sensitivity C-reactive protein (hsCRP) levels and a significant upregulation of the EGR1-TNF-α axis, creating a distinct, severe pro-inflammatory signature 93335. Furthermore, ASXL1 mutations alter systemic metabolic signaling, interfering fundamentally with mitochondrial function and energy homeostasis in cardiac tissue. This distinct metabolic and transcriptomic disruption explains why ASXL1 mutations, unlike DNMT3A, are highly correlated with reduced left ventricular ejection fraction (LVEF) and a rapidly progressing, non-ischemic heart failure phenotype in experimental models 3435.
5.3 The Thrombotic Imperative: JAK2 V617F
The JAK2 V617F mutation, a gain-of-function alteration classically defining myeloproliferative neoplasms such as polycythemia vera and essential thrombocythemia, acts as a uniquely potent driver of cardiovascular disease even at the lower variant allele frequencies characteristic of CHIP 3637. The pathophysiological mechanisms underpinning the cardiovascular risk of JAK2 CHIP are distinct, multifaceted, and predominantly thrombotic rather than purely atherosclerotic:
- Arterial Thrombosis via Platelet Hyperreactivity: JAK2-mutant megakaryocytes in the bone marrow exhibit elevated proplatelet formation, shedding hyperreactive, reticulated (young) platelets into the circulation. These young, enlarged platelets exhibit a massive, two- to three-fold upregulation of cyclooxygenase enzymes (COX-1 and COX-2) and cytosolic phospholipase A2 (cPLA2), leading to immense Thromboxane A2 (TXA2) synthesis 36. Critically, this overproduced mutant TXA2 acts in a paracrine manner as a potent amplifying signal, directly activating surrounding wild-type platelets and creating a cascading, systemic prothrombotic environment in the peripheral blood 36.
- Venous Thromboembolism (VTE) and Neutrophil Priming: Independently of arterial pathways, JAK2 mutations profoundly alter leukocyte biology by priming circulating neutrophils to undergo spontaneous NETosis - the explosive release of neutrophil extracellular traps (NETs). These ejected webs of chromatin, DNA, and histones act as a highly thrombogenic biological scaffold, trapping red blood cells and platelets and significantly elevating the risk of deep vein thrombosis and life-threatening pulmonary embolism 3637. Furthermore, JAK2 clones activate the AIM2 inflammasome, a DNA-sensing complex distinct from NLRP3, which further drives the necrotic and thrombotic deterioration of existing vascular lesions 810.
6. Expanding the Cardiometabolic Phenotype: Beyond Ischemia
The initial clinical recognition of CHIP focused almost exclusively on traditional atherosclerotic endpoints, particularly myocardial infarction and ischemic stroke 28. However, comprehensive research spanning 2024 to 2026 has unequivocally established CHIP as a multi-system cardiometabolic driver, implicating these mutant clones directly in the pathogenesis of progressive, non-ischemic heart diseases and providing a molecular rationale for age-related cardiac decline.
6.1 Heart Failure with Preserved Ejection Fraction (HFpEF)
Heart Failure with Preserved Ejection Fraction (HFpEF) is increasingly recognized not merely as a localized hemodynamic failure, but as a chronic, systemic inflammatory syndrome driven by metabolic dysfunction, obesity, hypertension, and aging - making its pathophysiology inextricably linked to CHIP 1138.
Recent high-impact clinical data drawn from the UTSW HFpEF cohort and the TOPCAT trial explicitly demonstrate that the presence of CHIP - particularly TET2 mutations - is an independent risk factor for incident HFpEF, increasing the longitudinal risk of heart failure hospitalization and all-cause mortality by over 2.3-fold (HR 2.35) independently of existing coronary artery disease 11133148.
Mechanistically, 2025 murine models designed to replicate cardiometabolic HFpEF (utilizing high-fat diets and hypertension induction) reveal that disease progression prompts profound extramedullary hematopoiesis, marked by elevated circulating CD34+ hematopoietic stem cells 1138. The resulting elevated fatty acid metabolism inside mutant macrophage mitochondria directly remodels the Vcam1 promoter, enhancing cellular adhesion and driving a systemic, smoldering inflammation that ultimately results in severe cardiac diastolic dysfunction and interstitial fibrotic remodeling, entirely independent of acute coronary ischemia 113138.
6.2 Arrhythmogenesis and Atrial Fibrillation (AF)
Emerging electrophysiological data suggest a robust, independent association between CHIP and new-onset cardiac arrhythmias 39. CHIP carriers exhibit a significantly increased propensity for incident AF 324. Similar to the findings in HFpEF, TET2 mutations appear to confer the highest genetic risk for the development of AF 2432. The chronic, low-grade inflammatory state generated by circulating mutant leukocytes - specifically propagated via the NLRP3 inflammasome axis - drives structural and electrical remodeling of the atrial myocardium, leading to severe left atrial enlargement, disrupted cellular electrophysiology, and subsequent, intractable arrhythmogenesis 2432.
6.3 Advanced Plaque Phenotyping via Artificial Intelligence
The structural consequences of CHIP-driven inflammation on coronary anatomy are being newly visualized through advanced imaging techniques. The late-breaking FISH&CHIPS study, presented at the AHA Scientific Sessions in 2025, utilized AI-driven coronary computed tomography angiography (CCTA) on nearly 8,000 patients to demonstrate that total plaque volume (TPV) is a powerful, independent predictor of long-term cardiovascular events . When CHIP mutations coincide with high LDL cholesterol, serial CCTAs reveal a measurable, rapid early impact on coronary arteries, characterized by the accelerated emergence of new, highly unstable necrotic plaques in proximal lesions 25. This confirms that CHIP operates synergistically with traditional metabolic factors to actively destabilize the vascular wall 25.
7. The Clinical Debate: Screening, Risk Stratification, and Guidelines
As CHIP incontrovertibly solidifies its status as a potent, modifiable cardiovascular risk factor, the clinical community faces urgent, highly debated questions regarding optimal screening protocols, dynamic risk stratification, and the initiation of pharmacological intervention in asymptomatic carriers.
7.1 Routine Population Screening vs. Targeted Incidental Assessment
Currently, leading global cardiology societies - including the American College of Cardiology (ACC) and the American Heart Association (AHA) in their updated 2026 guidelines - do not recommend broad, population-level genomic screening for CHIP 337404142. The core arguments against universal screening point to the current lack of proven, FDA-approved therapies specifically indicated to reverse CHIP, the psychological burden of diagnosing an otherwise healthy individual with a "pre-leukemic" state, and the epidemiological reality that the majority of individuals harboring small clones (VAF <10%) will never experience adverse cardiovascular or oncologic outcomes 3437.
However, the clinical paradigm is actively shifting from population screening toward rigorous targeted assessment 8. CHIP is now frequently detected incidentally during genetic testing for solid tumors, hereditary cancer panels, liquid biopsies, or the routine evaluation of mild, unexplained cytopenias 3843. Consequently, specialized, multidisciplinary "CHIP Clinics" and integrated Cardio-Oncology centers are emerging globally to manage this unique patient population 81143.
For patients with an established diagnosis of CHIP, cardiovascular management strategies must pivot to become highly aggressive. The presence of high-risk CHIP features - defined as a VAF >10%, the presence of multiple concurrent mutations, or the detection of high-risk alleles such as JAK2 and ASXL1 - warrants the immediate categorization of the patient into an elevated cardiovascular risk tier 8394144. This necessitates strict adherence to guideline-directed medical therapy, encompassing intensive lipid lowering (e.g., maximizing statin dosages, early initiation of PCSK9 inhibitors), uncompromising blood pressure control, and vigilant, serial monitoring for subclinical atherosclerosis utilizing non-contrast coronary artery calcium (CAC) scoring 8394144.
7.2 The "Incident vs. Recurrent" Paradox in Established CVD
A critical, deeply nuanced shift in CHIP risk stratification emerged from a massive 2024 observational analysis encompassing 63,700 patients across five major TIMI randomized clinical trials 4557. The study aimed to evaluate whether the presence of CHIP modulated the ultimate clinical benefit of advanced cardiovascular therapies (including PCSK9 inhibitors, SGLT2 inhibitors, and novel antiplatelet agents) in patients with established ASCVD.
The findings were paradigm-altering for cardiovascular risk models: while the presence of CHIP was strongly associated with a 30% increased risk of experiencing a first myocardial infarction in primary prevention cohorts, it was decidedly not associated with an increased risk of recurrent cardiac events in secondary prevention populations who were already receiving intensive, modern medical therapy 4557. Furthermore, patients harboring CHIP derived the exact same relative magnitude of benefit from standard, advanced ASCVD therapies as those without CHIP, with no outsized, mutation-specific benefit observed for broad-spectrum agents 4557. This paradox suggests a vital biological reality: once atherosclerosis is highly advanced, structurally established, and aggressively treated pharmacologically, the relative contribution of CHIP-driven inflammation to subsequent plaque rupture may be fundamentally blunted. Consequently, the highest clinical utility for CHIP screening and early intervention lies decisively in primary, rather than secondary, cardiovascular prevention 4557.
8. Precision Therapeutics and Future Directions
The precise mechanistic elucidation of the CHIP-inflammatory axis has catalyzed intense interest in both repurposed anti-inflammatory drugs and novel, targeted therapeutics aimed at suppressing clone-specific pathways, marking the dawn of precision cardiovascular medicine for age-related decline.
8.1 Targeting the IL-1β / IL-6 Axis
The central role of the inflammasome in TET2 and DNMT3A CHIP has made the IL-1β to IL-6 pathway the primary target for pharmacological interception. * IL-1β Inhibition: Retrospective, post-hoc analyses of the landmark CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcomes Study) trial revealed that individuals harboring TET2 CHIP mutations derived an "outsized, highly significant benefit" from the IL-1β neutralizing monoclonal antibody canakinumab, experiencing substantially lower rates of major adverse cardiovascular events and heart failure hospitalizations compared to non-CHIP carriers receiving the same therapy 13323337. * IL-6 Inhibition: Given that IL-1β acts upstream to drive systemic IL-6 production, direct IL-6 blockade is under intense clinical investigation. Human Mendelian randomization studies confirm that genetically reduced IL-6 signaling safely attenuates CVD risk specifically in carriers of large DNMT3A or TET2 clones, without the concurrent risk of severe infection seen with broad IL-1β blockade 334646. Novel monoclonal antibodies directly targeting IL-6, such as ziltivekimab, are currently advancing through late-stage development to determine their precise efficacy in mitigating high-inflammatory ASCVD phenotypes driven by CHIP 10. The ENTRACTE trial has additionally demonstrated the cardiovascular safety of existing IL-6 receptor inhibitors like tocilizumab, paving the way for targeted cardiovascular trials 4646.
8.2 Inflammasome Modulation and Colchicine
The LoDoCo2 (Low-Dose Colchicine) trial substudy, presented prominently in late 2025, provided remarkable evidence that low-dose colchicine - an inexpensive, orally available, and widely utilized anti-inflammatory agent - can significantly blunt the longitudinal growth trajectories of certain CHIP clones, particularly TET2 mutations 17. By dampening the inflammatory feedback loop that provides mutant stem cells with a selective survival advantage, colchicine fundamentally alters the bone marrow niche. Furthermore, colchicine therapy substantially reduced circulating IL-6 levels in patients with non-DNMT3A driven CHIP, offering a highly accessible, immediately available avenue for targeted cardiovascular and oncologic prevention in clonal populations 17.
8.3 Metabolic and Epigenetic Interventions
Beyond broad anti-inflammatory strategies, trials are increasingly investigating therapies that directly target the epigenetic machinery disrupted by CHIP mutations. High-dose oral and intravenous ascorbic acid (Vitamin C) is currently in clinical trials (NCT03682029, NCT03418038) for TET2-mutant CCUS, as ascorbic acid acts as an essential cofactor for TET enzymes and may restore residual epigenetic function to mutant cells 16. Similarly, targeted IDH1 and IDH2 inhibitors, such as ivosidenib and olutasidenib, initially approved for acute myeloid leukemia, are now being deployed in clinical trials (NCT05030441, NCT06566742) for high-risk CCUS patients, attempting to halt leukemogenic progression and cardiovascular damage at the earliest stages of clonal expansion 16.
9. Conclusion
Clonal hematopoiesis of indeterminate potential represents a watershed biological discovery that permanently bridges the formerly disparate disciplines of hematology, gerontology, and cardiovascular medicine. Moving decisively away from its narrow historical classification as merely an oncologic precursor state, the robust 2024 to 2026 research landscape firmly establishes CHIP as a primary, causal driver of systemic cardiometabolic decline and premature mortality.
The cardiovascular sequelae of CHIP are not uniform; they are dictated by precise genetic architecture and ancestral variables. Epigenetic modifiers like TET2 and DNMT3A orchestrate massive inflammasome-driven atherogenesis and HFpEF; ASXL1 variants induce profound metabolic derangements, distinct proteomic signatures, and rapid heart failure progression; and JAK2 mutations fundamentally alter platelet rheology and neutrophil biology to induce severe arterial and venous thrombosis. Furthermore, as diverse global biobanks from the "All of Us" program to Asian and Latin American registries continuously highlight, the baseline prevalence, dominant mutation types, and specific phenotypic threats of these clones vary significantly across geographic and racial lines, necessitating culturally and genetically tailored approaches to risk assessment.
While broad, untargeted population screening is not yet endorsed by major cardiological societies due to the lack of approved curative therapies, the incidental discovery of CHIP demands immediate clinical recognition as a profound risk-enhancing factor. Future breakthroughs in the management of human aging rely entirely on the successful execution of precision cardiology trials - such as those actively evaluating targeted IL-6 inhibitors, targeted inflammasome modulators like colchicine, and epigenetic restorers - to determine if mutational profiling can ultimately guide bespoke, life-extending therapeutic strategies for the aging cardiovascular system.