Circadian rhythm disruption and restoration in aging
The mammalian circadian timing system is a fundamental evolutionary adaptation that synchronizes physiological, behavioral, and metabolic processes with the 24-hour solar cycle. As organisms age, this highly calibrated system undergoes profound structural and functional deterioration, leading to a progressive loss of temporal homeostasis. Circadian disruption in aging is characterized by dampened rhythmic amplitude, altered phase timing, and a decoupling of central and peripheral clocks. These temporal alterations do not merely accompany chronological aging; they are actively implicated in accelerating the biological hallmarks of aging, including cellular senescence, disabled macroautophagy, and metabolic dysfunction. Conversely, interventions that restore circadian integrity - such as timed light exposure, chrononutrition, and pharmacological resynchronization - demonstrate significant potential to mitigate age-related decline, preserve physiological function, and extend healthspan.
Evolutionary and Molecular Architecture of Circadian Clocks
To comprehend how aging dismantles circadian regulation, it is necessary to examine the hierarchical network that governs biological timekeeping, its evolutionary origins, and the molecular mechanisms that drive it.
Geographic Adaptation and Seasonal Variation
Modern humans are believed to have originated near the equator in Africa, a region characterized by a nearly constant 12-hour light and 12-hour dark cycle throughout the year 1. As human populations migrated to higher latitudes, they encountered profound seasonal variations in photoperiods 1. Genomic analyses reveal that natural selection acted heavily on circadian regulatory mechanisms during this expansion. Differences in specific alleles across 84 genes known to regulate circadian rhythms and sleep cycles have been identified in populations living at extreme latitudes 1. This genetic adaptation suggests that tuning the biological clock to environmental light variations was critical for survival and reproductive success 1.
In contemporary cohorts, this latitudinal impact remains observable. Data from large-scale human tracking indicates that individuals in temperate regions (above the 50th parallel north) average roughly 7.3 hours of sleep per night with 90% sleep efficiency, whereas those in equatorial regions (below the 15th parallel north) average 6.6 hours with 88.5% efficiency 2. In high-latitude environments, such as polar regions experiencing continuous winter darkness or continuous summer daylight, the lack of a stabilizing photic zeitgeber routinely induces circadian phase delays, desynchronization, and subsyndromal seasonal affective alterations 3.
The Master Pacemaker and Peripheral Oscillators
The central circadian pacemaker resides in the suprachiasmatic nucleus (SCN) of the anterior hypothalamus 456. Comprising tens of thousands of neurons, the SCN receives direct photic input from intrinsically photosensitive retinal ganglion cells (ipRGCs) via the retinohypothalamic tract 578. Light serves as the primary zeitgeber (time cue), entraining the SCN to the external 24-hour day 7910. The SCN, in turn, orchestrates systemic rhythmicity by transmitting neuroendocrine and autonomic signals - such as rhythmic cortisol and melatonin secretion - to peripheral clocks distributed throughout virtually every tissue and organ system, including the liver, skeletal muscle, heart, and gastrointestinal tract 511121314.
While the SCN is indispensable for maintaining systemic synchrony and driving behavioral rhythms like sleep and wakefulness, peripheral tissues possess their own self-sustained circadian oscillators 111415. For instance, the liver clock regulates up to 15% of the hepatic transcriptome, governing critical metabolic pathways such as glucose homeostasis, xenobiotic detoxification, and lipid metabolism 5811. These peripheral clocks are highly responsive to non-photic zeitgebers, most notably feeding-fasting cycles and nutrient availability. Consequently, systemic temporal harmony relies on the continuous, synchronized dialogue between the light-entrained SCN and the nutrient-entrained peripheral tissues 5111316.
The Molecular Transcription-Translation Feedback Loop
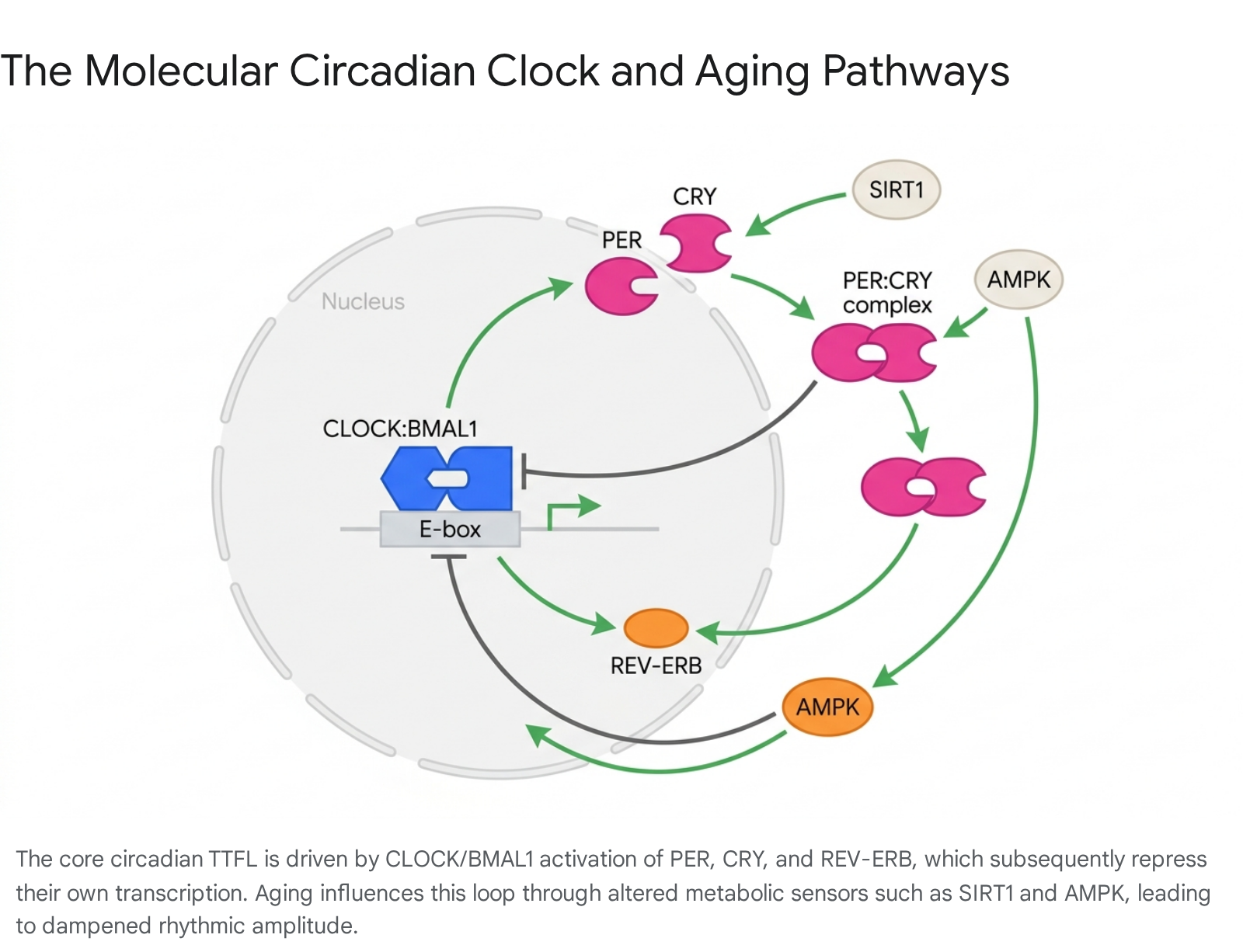
At the cellular level, circadian rhythms are generated by an autoregulatory transcription-translation feedback loop (TTFL) 561216. The core, or positive limb, of this loop consists of the basic helix-loop-helix transcription factors CLOCK (Circadian Locomotor Output Cycles Kaput) and BMAL1 (Brain and Muscle ARNT-Like 1) 51213. The CLOCK:BMAL1 heterodimer binds to E-box enhancer elements in the promoters of target genes, driving the transcription of the negative limb components, specifically the Period (Per1, Per2, Per3) and Cryptochrome (Cry1, Cry2) genes 121617.
Once translated and accumulated in the cytoplasm, PER and CRY proteins form complexes that translocate back into the nucleus to repress the transcriptional activity of CLOCK:BMAL1, thereby halting their own transcription 1216. This repression is eventually relieved through the phosphorylation and subsequent proteasomal degradation of PER and CRY proteins by kinases such as Casein Kinase 1 delta/epsilon (CK1δ/ε) and AMP-activated protein kinase (AMPK) 1218.
An auxiliary stabilizing loop involves the nuclear receptors REV-ERB (α and β) and ROR (α, β, and γ), which compete for binding at retinoic acid-related orphan receptor response elements (ROREs) to respectively repress and activate Bmal1 transcription 1219. The precise temporal delay built into this TTFL results in an oscillation that cycles approximately every 24 hours, regulating a vast array of clock-controlled genes that govern local cellular physiology 121620.
Age-Related Alterations in Circadian Rhythmicity
The aging process universally compromises the integrity of the circadian timing system. This deterioration manifests physiologically as advanced chronotype (a shift toward morningness), severely fragmented sleep-wake cycles, and flattened endocrine rhythms.
Dampening of Pacemaker Amplitude and Phase Advancement
Two of the most consistent age-related circadian changes are amplitude attenuation and phase advancement 4212223. Amplitude refers to the magnitude of the oscillation between the peak and trough of a circadian rhythm 16. In older adults, the amplitude of core body temperature rhythms decreases by 20% to 40%, failing to reach the low nadir necessary for consolidated restorative sleep 8. Similarly, the amplitude of endogenous melatonin secretion - the primary neurohormonal signal of subjective night - declines sharply, dropping by 40% or more in aging populations, while its peak shifts 1.5 to 2 hours earlier in the evening 82123.
This phase advance shifts the onset of physiological processes earlier in the 24-hour cycle, leading to early evening sleepiness and premature morning awakenings 82124. The mechanisms underlying these shifts are multifaceted. At the ocular level, progressive yellowing of the lens and a decline in the density of ipRGCs significantly restrict the transmission of short-wavelength (blue) light to the SCN, effectively depriving the pacemaker of its most potent entraining signal 8212526. Within the SCN itself, aging reduces the amplitude of electrical firing rhythms and diminishes the expression of critical coupling neuropeptides such as vasoactive intestinal polypeptide (VIP) and arginine vasopressin (AVP) 827.
Sleep Architecture Degradation and Homeostatic Shifts
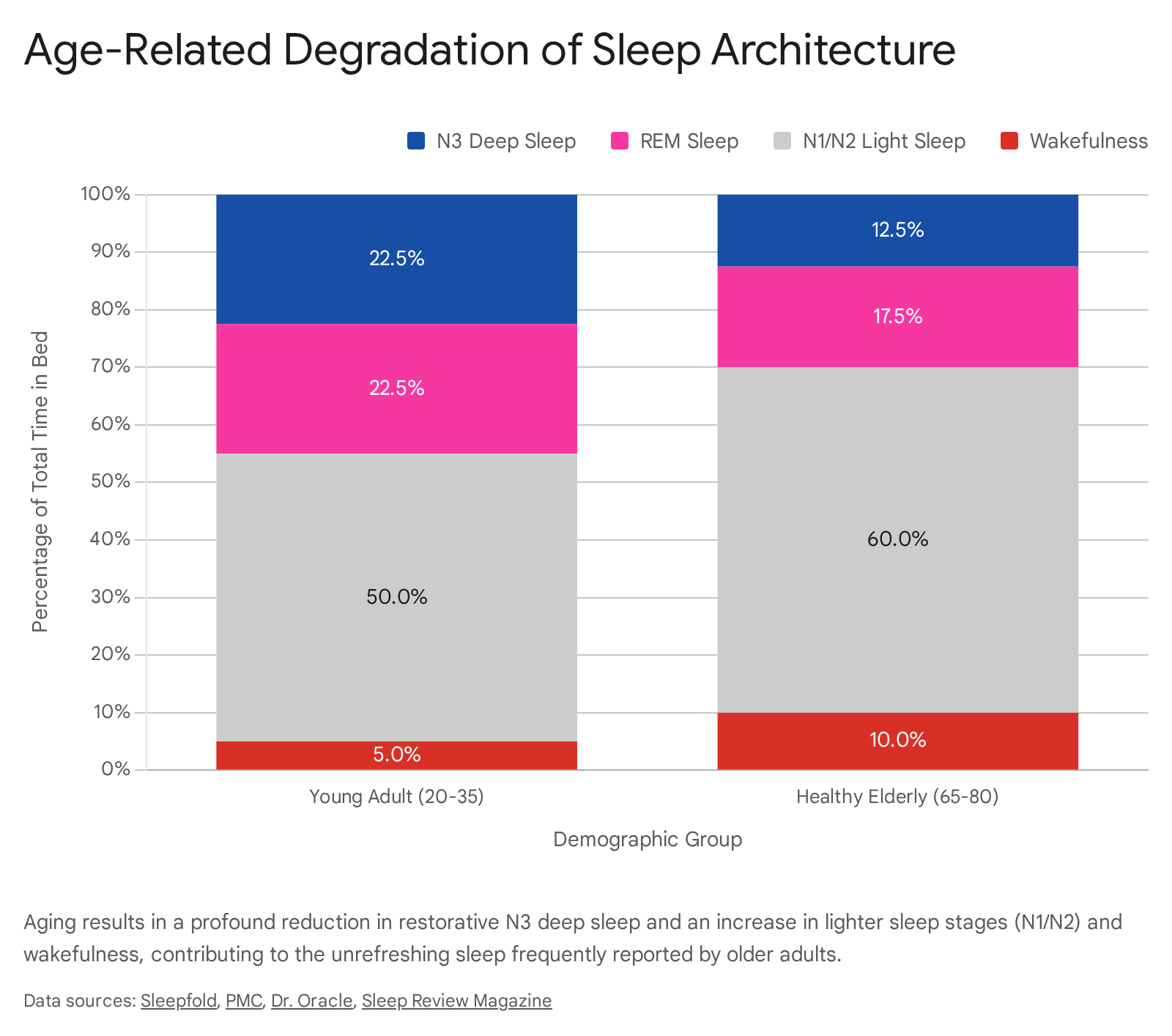
Sleep is the most prominent behavioral output of the circadian system, heavily regulated by the interaction between the circadian drive for wakefulness and the homeostatic accumulation of sleep pressure 42829. Aging profoundly degrades sleep architecture. Total sleep time decreases by approximately 10 minutes per decade until age 60, after which it stabilizes at a lower baseline 3031.
The most dramatic deterioration occurs in N3 (slow-wave or deep) sleep. N3 sleep is critical for glymphatic clearance of neurotoxic waste, metabolic restoration, and memory consolidation 2131. From early adulthood (where it constitutes roughly 20% to 25% of sleep) to midlife, N3 sleep declines precipitously, decreasing by roughly 8% per decade after the age of 30 2133. By the age of 70, many individuals experience a near-total absence of highly consolidated slow-wave activity, with corresponding prefrontal gray matter atrophy 21.
Rapid Eye Movement (REM) sleep is relatively preserved until the sixth decade of life but begins to decline by approximately 10 minutes per decade thereafter 33. Consequently, the sleep profile of older adults becomes dominated by lighter N1 and N2 sleep stages. These lighter stages are inherently unstable, leading to a massive increase in Wakefulness After Sleep Onset (WASO) and nocturnal awakenings, which can increase by over 150% compared to young adults 42133. The blunted homeostatic sleep drive in older adults also reduces the intensity of subsequent recovery sleep after periods of sleep deprivation 42829.
Table 1: Quantitative Age-Related Changes in Sleep Architecture
The following table summarizes the structural degradation of human sleep comparing healthy young adults to healthy elderly populations.
| Sleep Parameter | Young Adult (20-35 years) | Healthy Elderly (65-80 years) | Trajectory / Change |
|---|---|---|---|
| Total Sleep Time (TST) | 7.0 - 8.0 hours | 6.0 - 7.0 hours | Decreases ~10 min per decade |
| N3 Deep Sleep (%) | 20% - 25% | 10% - 15% (often lower) | Declines ~8% per decade |
| REM Sleep (%) | 20% - 25% | 15% - 20% | Declines late in life |
| N1 & N2 Light Sleep (%) | ~50% | >60% | Progressive increase |
| Sleep Onset Latency | 10 - 20 minutes | 20 - 40 minutes | Increases by up to 100% |
| Nighttime Awakenings | 1 - 2 | 3 - 5 | Increases by >150% |
| Sleep Efficiency | > 90% | 75% - 85% | Drops below 85% |
Data compiled from quantitative polysomnographic reviews of healthy aging cohorts 21303332.
Desynchronization Between Central and Peripheral Clocks
Perhaps more detrimental than the isolated dampening of the SCN is the ensuing internal desynchronization across the organism. The aging circadian network suffers from uncoupling, where peripheral tissues lose coordination with the master pacemaker and with each other 81119.
In vivo studies using PER2::LUCIFERASE reporter mouse models indicate that while the aged SCN may retain baseline oscillatory capacity (albeit with an age-related shortening of its free-running period), peripheral tissues such as the lung, kidney, pineal gland, and thymus undergo pronounced phase-advances and expression dampening 82733. Crucially, when subjected to environmental perturbations - such as a 6-hour phase advance modeling transmeridian travel or shift work - peripheral tissues in aged mice exhibit severely slowed rates of re-entrainment compared to young mice, taking days longer to re-align their core clock genes 833.
Because peripheral clocks govern tissue-specific metabolic processes, this loss of temporal hierarchy has severe systemic implications. The liver, for example, relies on precise temporal gating to separate mutually exclusive biochemical processes (e.g., glycolysis versus gluconeogenesis) 722. When the liver clock is desynchronized from the SCN - due to an aging-related loss of robust neuroendocrine signals or chaotic dietary patterns - metabolic homeostasis breaks down, driving insulin resistance, dyslipidemia, and systemic frailty 781119. Furthermore, multi-omics analyses of skeletal muscle from aged non-human primates show that this desynchronization correlates with an accumulation of maladaptive lipids (such as palmitoylcarnitine) and disrupted fatty-acid beta-oxidation, accelerating the onset of sarcopenia 34.
The Intersection of Circadian Disruption and the Hallmarks of Aging
Circadian disruption is not merely a consequence of aging; it is an active driver of the aging process. Modern geroscience has codified a series of interconnected biological "hallmarks of aging," which have recently been expanded to twelve distinct categories, including genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, disabled macroautophagy, deregulated nutrient-sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, chronic inflammation, and dysbiosis 3536373839. Extensive evidence indicates a reciprocal, tightly coupled relationship between the circadian clock and several of these core aging mechanisms.
Cellular Senescence and Bidirectional Circadian Decline
Cellular senescence is a state of irreversible cell cycle arrest accompanied by a pro-inflammatory senescence-associated secretory phenotype (SASP) 404142. Senescent cells accumulate with age, secreting cytokines and proteases that degrade local tissue function and drive systemic "inflammaging" 64041.
Research indicates that cellular senescence fundamentally alters the endogenous circadian clock. Human primary fibroblasts induced into replicative senescence, or subjected to oxidative stress via hydrogen peroxide, exhibit profound circadian changes: their circadian period significantly lengthens, their phase is delayed, and the amplitude of core clock genes (PER1, PER2, CRY1) is severely downregulated 174243. This demonstrates that the accumulation of senescent cells directly degrades tissue-level circadian rhythmicity 4243.
Conversely, targeted disruption of the circadian clock rapidly accelerates senescence and aging 20. Mice deficient in Bmal1 (BMAL1-knockout models) exhibit a devastating premature aging phenotype characterized by a drastically reduced lifespan, rapid onset of sarcopenia, severe cataracts, loss of subcutaneous fat, ectopic calcification of tendons, and male and female sterility 121420. The BMAL1-deficient tissue shows highly elevated markers of oxidative stress and rapid accumulation of senescent cells, proving that a functional molecular clock is essential for suppressing the hallmarks of aging 121320. Interestingly, mutations in the Clock gene lead to a less severe phenotype (primarily presenting as dermatitis and cataracts), suggesting that different components of the TTFL play distinct non-redundant roles in genotoxic stress responses 20.
Disabled Macroautophagy and Loss of Proteostasis
Disabled macroautophagy is recognized as a primary hallmark of biological aging 39444546. Autophagy is the cellular quality control process that sequesters and recycles damaged organelles, misfolded proteins, and toxic aggregates 4446. With advancing age, autophagic flux declines, leading to a loss of proteostasis and an accumulation of cellular waste that underlies neurodegenerative diseases and metabolic decline 4446.
The circadian clock intimately regulates macroautophagy. Key nutrient-sensing pathways that control autophagy, such as the mechanistic target of rapamycin (mTOR) and AMPK, are directly intertwined with the circadian TTFL 104447. Under healthy conditions, the clock enforces rhythmic waves of autophagy - typically peaking during fasting and sleep phases - to clear cellular debris accumulated during the active phase 4748. As circadian amplitude dampens with age, this rhythmic induction of autophagy is severely impaired 1247. The consequent failure to clear damaged mitochondria and misfolded proteins creates a toxic intracellular environment that further suppresses clock gene expression, creating a destructive self-reinforcing feedback loop 12.
Mitochondrial Dysfunction and Oxidative Stress
Mitochondria, the primary source of cellular ATP, generate reactive oxygen species (ROS) as a metabolic byproduct. Mitochondrial dysfunction results in elevated oxidative stress that damages DNA, proteins, and lipids, contributing heavily to ischemic and neurodegenerative pathologies 6363840. The circadian clock actively regulates mitochondrial dynamics, including fission, fusion, and the synthesis of NAD+ (nicotinamide adenine dinucleotide), a crucial coenzyme for mitochondrial energy metabolism 10254049.
In aged tissues, the loss of circadian regulation drastically reduces the rhythmic biosynthesis of NAD+, starving mitochondria of energy and regulatory capacity 1049. This age-dependent decline in NAD+ impairs the activity of SIRT1, an NAD+-dependent deacetylase that functions as an accessory clock protein linking energy status to circadian phase 81040. SIRT1 regulates the acetylation of BMAL1 and PER2; as its activity drops with age, the resulting oxidative stress feeds back into the clock machinery, degrading clock proteins and perpetuating the loss of circadian fidelity 840. In cardiac tissue, researchers have demonstrated that boosting NAD+ via precursors like nicotinamide riboside (NR) successfully reprograms the cardiac circadian clock, restoring rhythmic gene expression and reversing age-related cardiac enlargement 1249.
Inflammaging and Immune Dysregulation
Chronic, low-grade inflammation in the absence of infection - termed "inflammaging" - is a critical driver of frailty and chronic disease 4550. Immune cells, particularly macrophages, possess robust intrinsic clocks that govern their cytokine secretion and phagocytic activity 5152.
Recent studies have identified that the transcription factor Krüppel-like factor 4 (KLF4) acts as a key mediator in the circadian regulation of immune responses 52. In aged macrophages, KLF4 abundance and rhythmicity diminish despite a functional core molecular clock, leading to a loss of circadian immune control and elevated basal inflammation 52. Furthermore, profiling of organismal aging trajectories via the "Digital Aging Twin" framework has identified that age-driven accumulation of liver-derived coagulation factors (particularly F13B, F9, and F10) acts as a direct driver of systemic vascular aging 53. When these factors lose strict temporal gating, they prompt endothelial cells to exhibit senescence, impaired tube formation, and elevated inflammatory signaling 53.
The Gut-Brain-Muscle Axis in Circadian Aging
The gastrointestinal tract represents one of the largest peripheral circadian organs, and its temporal regulation is intricately tied to the trillions of microorganisms residing within it. The gut microbiome exhibits its own diurnal oscillations in composition, localization, and metabolic output, all of which are highly sensitive to chronological aging and circadian disruption 135455.
Microbiome Rhythms and Age-Related Dysbiosis
Healthy gut microbiota undergo distinct 24-hour cycles of abundance, driven heavily by the host's feeding-fasting patterns and the intrinsic circadian clock of the intestinal epithelium, which regulates barrier renewal and antimicrobial peptide production 135657. As humans age, the overall diversity of the gut microbiome declines, leading to dysbiosis - an imbalance characterized by an overrepresentation of pro-inflammatory bacterial strains and a loss of beneficial populations 505457.
Chronodisruption - induced by erratic eating schedules, fragmented sleep, or decreased light exposure in older adults - exacerbates this age-related dysbiosis. The loss of rhythmic mucosal immune signaling and intestinal barrier integrity results in "leaky gut," allowing microbial endotoxins to breach the epithelial barrier 505658. This systemic circulation of endotoxins fuels inflammaging, further deteriorating peripheral clock function in organs like the liver and the brain via the vagus nerve 505556.
Metabolite Signaling via Short-Chain Fatty Acids
The bidirectional dialogue between the host clock and the microbiome relies heavily on microbial metabolites. Beneficial bacteria ferment dietary fibers into short-chain fatty acids (SCFAs) such as butyrate, propionate, and acetate 505455. SCFAs act as potent signaling molecules that link the gut to systemic circadian rhythms 1355.
Microbiome-derived SCFAs, alongside secondary bile acids like lithocholic acid (LCA) and tryptophan derivatives, entrain the hepatic and peripheral clocks by serving as epigenetic modifiers (e.g., via histone deacetylase inhibition) and by activating G-protein-coupled receptors (such as GPR41/43 and TGR5) and nuclear receptors like Farnesoid X receptor (FXR) and PPAR-gamma 135559. In aging, the loss of SCFA-producing bacteria deprives peripheral clocks of these crucial timing and metabolic cues, resulting in dampened circadian amplitude in the liver and promoting insulin resistance, adiposity, and hepatic steatosis 13555758.
Chronobiological Interventions and Rhythm Restoration
While aging inexorably degrades the circadian network, research demonstrates that reinforcing exogenous circadian zeitgebers can effectively bypass weakened endogenous pacemakers. Interventions targeting the chronobiological axis - ranging from controlled light exposure and meal timing to targeted pharmacotherapy - show high potential for restoring rhythmicity and mitigating age-related functional decline.
Photobiomodulation and Targeted Light Therapy
Given that light is the primary entraining signal for the SCN, timed light therapy is a leading non-pharmacological intervention for restoring circadian amplitude in older adults. Age-related ocular changes and highly sedentary, indoor lifestyles effectively plunge many elderly individuals into a state of "biological darkness" during the day, failing to provide the threshold intensity required to suppress daytime melatonin and anchor the circadian phase 32660.
Clinical trials demonstrate that bright light therapy (frequently administered at intensities of up to 10,000 lux) can significantly improve sleep efficiency, reduce nighttime awakenings, and ameliorate depressive symptoms in aging populations 26636162. A recent meta-analysis assessing light therapy for depressive symptoms in older adults found a moderate, statistically significant effect size (Hedges' g = 0.525), with white and bluish light yielding the most substantial efficacy 61. For older adults suffering from neurodegenerative conditions like Alzheimer's Disease and Related Dementias (ADRD), timed light exposure has been shown to consolidate rest-activity patterns and reduce severe evening agitation 2663. However, broader Cochrane reviews note that evidence certainty remains low for certain quality-of-life metrics, underscoring the need for standardized dosimetry 646566.
Recent chronobiological evidence points to the vital importance of light timing. While morning bright light advances the clock (combating delayed sleep phases), emerging laboratory studies indicate that afternoon light exposure (approximately 10 hours after the core body temperature nadir) may be highly effective at increasing the amplitude of the circadian rhythm in older adults, addressing the primary cause of sleep fragmentation 2167. Furthermore, photobiomodulation (PBM) using non-thermal red and near-infrared wavelengths (600 - 1100 nm) has demonstrated preclinical efficacy in directly enhancing mitochondrial cytochrome c oxidase activity, accelerating tissue repair, improving vascular function via nitric oxide availability, and protecting against age-related cardiac decline 2568.
Chrononutrition and Time-Restricted Eating
If light is the master cue for the central brain clock, food is the master cue for peripheral clocks. Chrononutrition investigates how the timing of energy intake aligns with circadian metabolic capacity 1369. Because the aged SCN loses its strict control over peripheral tissues, imposing rigid feeding schedules acts as a powerful exogenous synchronizer, rescuing metabolic rhythms even when the central clock is impaired 1156.
Time-Restricted Eating (TRE), which consolidates all caloric intake into a specific daily window during the active phase, profoundly benefits the aging trajectory 5134869. Analysis of large-scale nutritional cohorts (e.g., NHANES) reveals that consuming the final daily meal earlier in the evening (before 9:00 PM) is associated with a significantly lower risk of biological aging 48. Late-night eating forces the liver clock to process nutrients during its subjective rest phase, creating a circadian misalignment that suppresses critical nocturnal autophagy, exacerbates insulin resistance, and elevates inflammatory markers 485769. Interestingly, organ-specific aging clocks suggest the most favorable eating windows narrow to 3:00-5:00 PM for optimal cardiac health, and 5:00-7:00 PM for optimal hepatic health 48.
Furthermore, adherence to Mediterranean dietary patterns interacts closely with circadian genetics. Longitudinal studies following tens of thousands of women over 25 years found that individuals adhering strictly to Mediterranean diets showed a 23% reduced risk of all-cause mortality, driven largely by reductions in systemic inflammation and insulin resistance 70. Interestingly, the traditional Mediterranean "siesta" (midday nap) interacts directly with specific genetic profiles; in individuals carrying specific genetic variants across the TAS2R38, CLOCK, and FTO genes, regular napping appears to support metabolic health and protect against obesity, whereas the identical behavior in individuals lacking these genetic predispositions correlates with metabolic dysfunction 717273.
Melatonin Supplementation and Neuroprotection
Melatonin is fundamentally a chronobiotic hormone, but its physiological role extends far beyond sleep initiation. Secreted by the pineal gland in response to darkness, melatonin serves as a systemic timing signal, a powerful endogenous antioxidant, and an immunomodulator 23747576. Because endogenous melatonin amplitude drops by up to 40% in older adults, supplementation represents a direct restorative intervention to counteract age-related decline 217778.
Beyond resetting circadian phase, melatonin directly penetrates cellular and mitochondrial membranes 237578. Inside the mitochondria, melatonin and its metabolites (such as AFMK and AMK) neutralize reactive oxygen and nitrogen species at their source, preventing oxidative damage to mitochondrial DNA and rescuing the mitochondrial dysfunction characteristic of aging 237879. Clinical and preclinical data indicate that exogenous melatonin upregulates antioxidant enzymes (e.g., superoxide dismutase, glutathione peroxidase) and blunts inflammaging by downregulating pro-inflammatory cytokine cascades 757678. Furthermore, in models of Alzheimer's and Parkinson's disease, melatonin administration reduces amyloid-beta toxicity, preserves synaptic plasticity, and slows cognitive decline, positioning it as a potent neuroprotective agent in aging populations 23767879.
Emerging Pharmacotherapy: NAD+ Precursors and Senolytics
Modern geroscience is increasingly turning to pharmacological targets that interface with both the hallmarks of aging and the circadian clock. The decline in NAD+ disables the clock's ability to track metabolic energy. Supplementation with NAD+ precursors, such as nicotinamide riboside (NR), has been shown in aged animal models to successfully reprogram the cardiac circadian clock, restore rhythmic gene expression, and reverse age-related cardiac hypertrophy 1249.
Finally, interventions utilizing senolytics (compounds that selectively clear senescent cells) are showing promise in restoring tissue homeostasis. In preclinical models, intermittent dosing of senolytics like Dasatinib + Quercetin, or Fisetin, has successfully reduced the senescent cell burden in multiple tissues, thereby lowering the SASP inflammatory load that degrades local circadian function 80. By combining chronotherapy (light and diet) with gerotherapeutics (mTOR inhibitors and senolytics), it may be possible to re-establish the biological rhythms of life, thereby extending healthspan and delaying the onset of age-related pathologies 3880.