Cellular senescence in the brain and neurodegeneration
Fundamentals of Cellular Senescence in the Central Nervous System
Cellular senescence represents a highly regulated, complex state of stable cell-cycle arrest accompanied by profound metabolic, chromatin, and secretory reprogramming. Originally conceptualized over half a century ago as an in vitro barrier to unlimited cellular proliferation - a phenomenon referred to as replicative exhaustion or the Hayflick limit - senescence is now widely recognized as a fundamental biological mechanism that operates in vivo across various tissues, including the central nervous system (CNS) 12. Within the brain, cellular senescence affects both proliferation-competent glial cells and post-mitotic neurons, fundamentally altering the tissue microenvironment and driving the progressive decline in cognitive and motor functions associated with physiological aging and age-related neurodegenerative diseases 345.
The initiation and enforcement of cellular senescence are primarily governed by two canonical tumor-suppressor pathways: the p53/p21CIP1 signaling axis and the p16INK4a/retinoblastoma (Rb) pathway 46. These pathways activate in response to a myriad of intrinsic and extrinsic cellular stressors. Prominent triggers include telomere attrition, chronic oxidative stress resulting in the excessive generation of reactive oxygen species (ROS), mitochondrial dysfunction, and the persistent activation of the DNA damage response (DDR) 5789.
In the specific context of the central nervous system, the accumulation of neurotoxic misfolded proteins, such as amyloid-beta (Aβ) oligomers, hyperphosphorylated tau, and alpha-synuclein, serve as potent initiators for stress-induced premature senescence (SIPS) in neighboring cells 310. Once the senescence program is initiated, affected cells undergo distinct morphological and biochemical changes. These include cellular enlargement and flattening, an expansion of the lysosomal compartment, and a marked increase in the activity of senescence-associated beta-galactosidase (SA-β-gal) 111. Furthermore, senescent cells develop robust anti-apoptotic pathways, often referred to as senescent cell anti-apoptotic pathways (SCAPs), which allow them to resist programmed cell death and accumulate chronically within the aging brain 121314.
Transient Versus Chronic Senescence
A critical distinction in the biology of cellular senescence is the temporal duration of the senescent state and the subsequent capacity for immune-mediated clearance. Transient, or acute, senescence is a physiologically necessary program activated during embryonic development, tissue regeneration, and wound healing 29. In these developmental and regenerative contexts, transiently senescent cells produce a localized, short-term secretory response that guides tissue remodeling, prevents excessive fibrosis, accelerates extracellular matrix formation, and stimulates repair mechanisms. Following their functional contribution, these transiently senescent cells are rapidly recognized and cleared by the immune system, allowing for complete tissue resolution without lasting pathology 211.
Conversely, chronic senescence - which acts as the primary driver of pathological brain aging - arises when the accumulation of cellular stress severely exceeds the capacity for immune clearance 214. Age-related immunosenescence, characterized by a progressive decline in the functional efficiency of the innate and adaptive immune systems, results in a systemic failure to eradicate damaged cells 15. Consequently, senescent cells linger in the central nervous system indefinitely, shifting from a beneficial physiological mechanism to a maladaptive, pathological burden. These chronically present senescent cells perturb brain homeostasis through continuous, unrestrained paracrine signaling, laying the biochemical foundation for widespread neurodegeneration 914.
The Senescence-Associated Secretory Phenotype
The most deleterious consequence of chronic cellular senescence is the acquisition of the senescence-associated secretory phenotype (SASP). The SASP is a complex, highly heterogenous secretome composed of pro-inflammatory cytokines, chemokines, matrix metalloproteinases (MMPs), growth factors, and non-protein signaling molecules like reactive oxygen species and nitric oxide 3716. The relentless secretion of these factors actively modifies the local tissue microenvironment, contributing to the chronic, low-grade sterile inflammation often termed "inflammaging" 151718.
Transcriptional Regulation of the Secretory Phenotype
The production and specific composition of the SASP are meticulously regulated by several converging intracellular signaling cascades. The most conserved mechanism governing SASP expression is the nuclear factor kappa B (NF-κB) signaling pathway, which is heavily responsible for the transcription of primary pro-inflammatory SASP components, including interleukin-6 (IL-6) and interleukin-8 (IL-8) 71619. NF-κB is frequently activated alongside the CCAAT/enhancer-binding protein beta (C/EBPβ). Together, these transcription factors create an autocrine feedforward loop that continuously amplifies the inflammatory signal within the senescent cell, ensuring the sustained release of SASP factors 2021.
Upstream of NF-κB, the mammalian target of rapamycin (mTOR) and p38 mitogen-activated protein kinase (MAPK) pathways act as central regulators, integrating diverse cellular stress signals and stabilizing SASP-related mRNA transcripts to facilitate their translation 162022. Additionally, the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway has been identified as a critical innate immune sensor of cytosolic DNA, which is often aberrantly extruded from the nucleus or mitochondria in senescent cells. Research indicates that the cGAS-STING pathway operates in tandem with the IL-6-STAT3 axis to aggressively drive neuroinflammation in models of Alzheimer's disease, heavily influencing the overall severity and composition of the SASP 423.
Secondary Senescence and Extracellular Vesicles
The SASP does not strictly operate in an autocrine manner; it functions as a potent paracrine mechanism that facilitates the spread of senescence to healthy, proliferating cells - a phenomenon recognized as secondary or bystander senescence 71524. Soluble cytokines, such as IL-1α and transforming growth factor-beta (TGF-β), are secreted into the extracellular space where they directly induce DNA damage, reactive oxygen species generation, and stress signaling in adjacent cells, forcing them into a senescence-like state 715.
Beyond the diffusion of soluble proteins, the SASP includes the robust release of small extracellular vesicles (sEVs), primarily exosomes and microvesicles, which function as critical structural vectors for the distal propagation of the senescence signal 182425. These senescence-associated exosomes (SA-EXOs) encapsulate specific bioactive cargos, including signaling proteins, lipid metabolites, and microRNAs (miRNAs). Profiling of exosomal SASP has identified marked alterations in structural membrane lipids, with significant increases in phosphatidylcholines, phosphatidylethanolamines, and sphingomyelins, indicative of broad cellular membrane dysregulation occurring during the transition into senescence 24.
Furthermore, SA-EXOs carry specific senescence-modulating microRNAs. For example, miR-146a and miR-9 are frequently packaged within these vesicles to modulate upstream activators of NF-κB (such as TRAF6 and IRAK1) and TGF-β pathways in recipient cells 151825. Additional miRNAs, including miR-34a and miR-23b, have been identified as drivers of aberrant lipid metabolism and bystander senescence 18. Through this mechanism, a single senescent cell, such as a localized astrocyte, utilizes both soluble cytokines and SA-EXOs to propagate secondary senescence outward. This paracrine signaling cascade ultimately induces secondary senescence in neighboring healthy microglia, oligodendrocyte progenitor cells, and endothelial cells, precipitating a breakdown of blood-brain barrier tight junctions, initiating synaptic loss, and triggering widespread microglial activation across the neurovascular unit 3712151826.
Non-Neuronal Senescent Cell Populations
While mature neurons are post-mitotic and possess unique mechanisms of biological aging (sometimes termed "neurescence," characterized by the atypical re-entry into the cell cycle and DNA damage), the glial and vascular compartments of the brain represent the primary, highly metabolically active reservoirs of classical cellular senescence 52728. Astrocytes, microglia, oligodendrocyte progenitor cells, and endothelial cells each exhibit distinct senescence trajectories and contribute to overall neurodegeneration through specialized pathological mechanisms.
Astrocytic Senescence
Astrocytes are the most abundant glial cells in the central nervous system, providing critical metabolic and trophic support to neurons, maintaining the integrity of the blood-brain barrier, and regulating synaptic transmission. With advancing chronological age, and particularly in response to neurotoxic aggregates like amyloid-beta and tau oligomers, astrocytes undergo profound cellular senescence 31029.
Senescent astrocytes display highly elevated markers of p53, p21, and p16, along with robust SA-β-gal activity and severe mitochondrial dysfunction characterized by aberrant fission and fusion dynamics 329. The transition into senescence results in a devastating loss of their homeostatic neuroprotective functions. Instead of buffering extracellular glutamate and providing metabolic substrates to neurons, senescent astrocytes become primary engines of chronic neuroinflammation. They secrete massive quantities of IL-6, IL-1β, and interferon-γ (IFNγ) 320. This highly toxic astrocytic SASP directly drives the hyperphosphorylation of tau and the subsequent deposition of neurofibrillary tangles in adjacent neurons, forming a pathological feedback loop that actively accelerates Alzheimer's disease progression 3.
Microglial Senescence
Microglia function as the primary resident macrophages of the brain, continuously surveilling the parenchyma for damage, pathogens, and cellular debris. While acute microglial activation is beneficial for tissue repair and amyloid clearance, chronic activation combined with age-related telomere erosion transforms microglia into a senescent, dystrophic state 71427.
Aged, senescent microglia - frequently exhibiting a transcriptomic overlap with the Disease-Associated Microglia (DAM) profile - exhibit severely impaired phagocytic capacity, preventing the effective clearance of misfolded proteins and apoptotic cells 273031. In models of tauopathy, p16-expressing senescent microglia accumulate densely around neurofibrillary tangles and secrete high levels of pro-inflammatory mediators, including IL-1α, TNF-α, C1q, and matrix metalloproteinases 3303132. The persistence of these senescent immune cells actively antagonizes the brain's regenerative capacity and perpetuates a highly destructive state of neuro-inflammaging.
Oligodendrocyte Progenitor Cell Senescence
Oligodendrocyte progenitor cells (OPCs) serve as the primary proliferative stem-like cells within the adult CNS. They are responsible for migrating, proliferating, and ultimately differentiating into mature oligodendrocytes to maintain and repair the myelin sheaths that insulate neuronal axons. Recent transcriptomic and pathological evidence has highlighted the unique susceptibility of OPCs to cellular senescence, particularly in the context of demyelinating diseases like progressive multiple sclerosis (PMS) and Alzheimer's disease 32233.
In demyelinated white matter lesions, OPCs are constantly subjected to oxidative stress and persistent inflammation, leading to terminal cell-cycle arrest and the widespread expression of senescence markers 22. Senescent OPCs fail to differentiate into mature myelinating oligodendrocytes, directly leading to progressive myelination failure. Furthermore, these senescent cells secrete a highly specific SASP, heavily featuring the release of High-Mobility Group Box 1 (HMGB1). Extracellular HMGB1 acts as a potent molecular inhibitor of OPC maturation, effectively locking neighboring, healthy progenitor cells in an undifferentiated state - a phenomenon formally termed the "senescence-stem lock model" 22. By preventing remyelination and actively suppressing local tissue plasticity, OPC senescence directly drives long-term cognitive and motor deterioration.
Endothelial Cell Senescence and the Blood-Brain Barrier
The blood-brain barrier (BBB) is a highly selective, semi-permeable boundary formed by brain microvascular endothelial cells (BMECs), pericytes, and astrocytic endfeet. It is essential for protecting the brain from systemic toxins and maintaining a strictly controlled biochemical microenvironment 834. Endothelial cell senescence is now recognized as a primary initiator of age-related BBB breakdown, a pathological event that frequently precedes observable cognitive decline and structural neurodegeneration 821.
Senescent BMECs exhibit significant morphological alterations, becoming enlarged and losing their tightly regulated junctional integrity. Specifically, premature senescence disrupts the proper plasma membrane localization of zonula occludens-1 (ZO-1), a critical tight junction protein. This delocalization leads to increased paracellular flux and general vascular leakiness 8. Additionally, senescent endothelial cells upregulate the expression of basement membrane-degrading proteases, such as MMP-2 and MMP-9, which physically dismantle the surrounding vascular architecture 821.
Recent discoveries have also implicated specific enzymes in the propagation of endothelial senescence. For example, 15-hydroxyprostaglandin dehydrogenase (15-PGDH) is significantly enriched in senescent BMECs during physiological aging and following traumatic brain injury. This enzyme actively participates in the deterioration of the BBB, facilitating the infiltration of systemic inflammatory factors, peripheral immune cells, and blood-borne pathogens into the brain parenchyma, thereby exacerbating the local neuroinflammatory response 343435.
| Cell Type | Canonical Senescence Markers | Primary SASP Components | Pathological Contribution to Neurodegeneration |
|---|---|---|---|
| Astrocytes | p16, p21, p53, SA-β-gal | IL-6, IL-1β, IFNγ, IL-8 | Exacerbates tau/Aβ accumulation; induces synaptic dysfunction via PSD-95 alterations. 32936 |
| Microglia | p16, p21, SA-β-gal | TNF-α, IL-1α, IL-6, C1q | Exhibits impaired phagocytosis; drives chronic neuroinflammation and DAM signatures. 273031 |
| OPCs | p16, p21, SA-β-gal | HMGB1, IL-1β, TNF-α | Fails to differentiate into oligodendrocytes; suppresses local stem cell niches ("senescence-stem lock"). 223337 |
| Endothelial Cells | p16, p21, γH2AX | MMP-2, IL-8, MCP-1 | Disrupts tight junctions (ZO-1); breaks down BBB, allowing systemic neurotoxins into the parenchyma. 82132 |
Mechanisms of Synaptic and Neuronal Damage
The ultimate, compounding consequence of cellular senescence in the brain is the physical and functional degradation of neural circuits. The SASP secreted by senescent glial and vascular cells does not merely cause generalized, non-specific inflammation; it biochemically targets highly specific molecular mechanisms governing synaptic transmission and structural plasticity in nearby neurons.
Post-Synaptic Density Alterations and Palmitoylation
A highly specific molecular mechanism by which the astrocytic SASP induces synaptic dysfunction involves the post-translational modification of synaptic scaffolding proteins. When astrocytes enter a state of senescence or are heavily activated by pathological insults (such as peripheral nerve injury, traumatic brain injury, or protein aggregates), they release immense quantities of IL-6 directly into the synaptic cleft 103839.
Elevated extracellular IL-6 signals neurons to significantly upregulate the expression of specific S-acyltransferases, primarily zDHHC2 and zDHHC3, which are located in the synaptosome and Golgi apparatus 3639. These specific enzymes are responsible for the palmitoylation of Postsynaptic Density Protein 95 (PSD-95), a critical scaffolding and anchoring protein located at excitatory glutamatergic synapses. The excessive, unchecked palmitoylation of PSD-95 artificially forces its prolonged, rigid accumulation at the postsynaptic membrane 364041.
This altered membrane trafficking directly drives the abnormal, excessive recruitment and tethering of ionotropic glutamate receptors, specifically the AMPA receptor subunit GluR1 and the NMDA receptor subunit NR2B 3839.
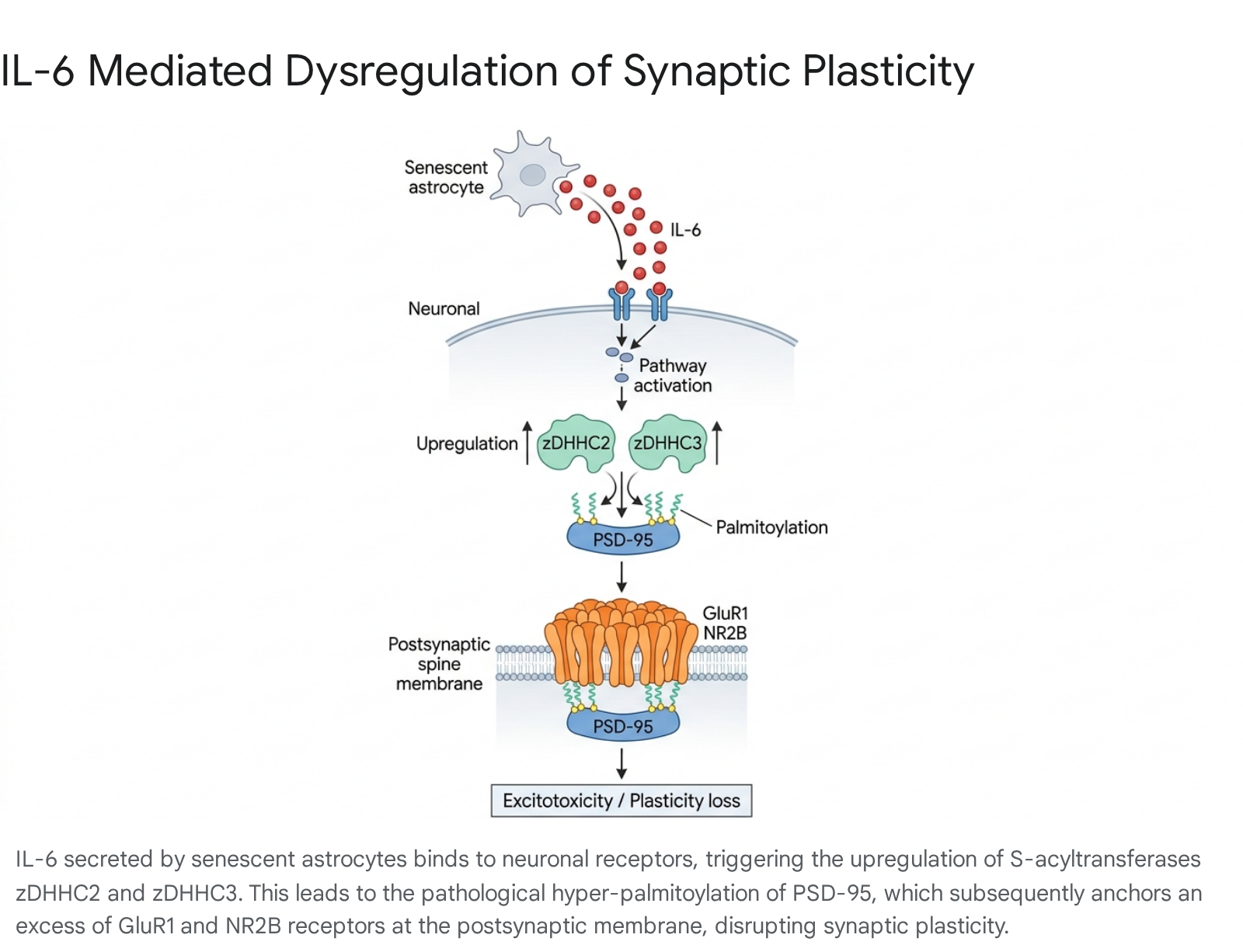
The dysregulation of glutamatergic synaptic composition disrupts homeostatic synaptic plasticity, leading to chronic excitotoxicity, the long-term depression of synaptic signaling, and significant cognitive and behavioral deficits. Experimental suppression of IL-6 or direct pharmacological inhibition of PSD-95 palmitoylation (via agents that modulate the depalmitoylating enzyme ABHD17) has been shown to effectively rescue these synaptic defects and restore normal learning and memory behavior in animal models 383940.
Extracellular Matrix Degradation by Matrix Metalloproteinases
Beyond direct receptor clustering, the structural and functional integrity of the synapse relies heavily on the extracellular matrix (ECM) and the specialized perisynaptic network. Senescent cells actively disrupt this delicate network through the hyper-secretion of matrix metalloproteinases, specifically MMP-3 1042.
In the CNS, MMP-3 does not merely degrade structural collagen or fibronectin; it acts directly on synaptic receptors. Following prolonged neuronal stimulation, MMP-3 is released into the extracellular space where it proteolytically cleaves the extracellular epitopes of the NMDA receptor NR1 subunit 43. Specifically, MMP-3 cleaves the NR1 subunit at the S542 and L790 sites, effectively shedding the glycine-binding domain of the receptor 43. This irreversible, activity-dependent alteration of the NMDA receptor structure directly impairs calcium influx and completely disrupts long-term potentiation (LTP), severing the brain's fundamental mechanism for learning and memory formation 424445. Uncontrolled, chronic MMP-3 activity is highly elevated in the brains of patients with Alzheimer's and Parkinson's diseases, marking it as a critical vector of SASP-mediated synaptic degradation 34246.
Transgenic Models and Detection Methods
The precise in vivo study of brain cellular senescence has historically been hindered by the lack of universally specific biomarkers, as well as the inherently low baseline abundance of senescence-related transcripts in healthy tissue. However, the development of highly sophisticated transgenic reporter and ablation mouse models has recently revolutionized the field, allowing for the direct mapping of senescent cells to neuropathology 13147.
Advances in Senescence Reporter Models
Initial attempts to track senescence utilized the p16-3MR (modality reporter) model. This mouse contains a fusion protein consisting of a synthetic Renilla luciferase, monomeric red fluorescent protein (mRFP), and truncated herpes simplex virus thymidine kinase (HSV-TK) under the control of the p16INK4a promoter. This design allowed for both the luminescent detection of senescent cells and their selective apoptotic clearance via the administration of the antiviral drug ganciclovir 131. However, recognizing potential off-target luminescence and unspecific signaling in certain tissues, newer models have been engineered with significantly higher precision.
The p16-tdTomato knock-in model integrates a bright fluorophore directly into the endogenous p16 locus, enabling the high-resolution visualization, isolation, and single-cell transcriptomic sequencing of rare senescent populations in the complex brain environment 3148. Similarly, the p16-CreERT2 system allows for the temporal, tamoxifen-inducible tracking of senescent cell lineages, providing insight into the fate of these cells over the organism's lifespan 3148.
Expanding beyond p16, novel p21-reporter models - such as the inducible p21-Cre mouse, the p21-ATTAC model, and the p21-3MR system - have provided crucial evidence that p16 and p21 characterize distinct, largely non-overlapping sub-populations of senescent cells, each driving highly customized SASP profiles 31. Innovative non-mammalian models have also been established to accelerate research, including the p21-GFP zebrafish and the fast-aging African Turquoise Killifish, alongside specialized reporter models in Drosophila (such as the AP1 reporter for senescent glia) 31.
Disease-Specific Senescence Trajectories
When these advanced reporter systems are crossbred with established models of neurodegeneration, they reveal a profound physiological chronology. In models like the MAPT-P301S (PS19) tauopathy mouse, the hTau mouse, and the App NL-G-F amyloid model, researchers observe that the accumulation of senescent glia occurs prior to the onset of widespread neurodegeneration and cognitive collapse 30323749. Furthermore, naturally fast-aging models, such as the Senescence-Accelerated Mouse-Prone 8 (SAMP8) and the OXYS rat strain, develop spontaneous cognitive deficits that closely parallel the accumulation of senescent cells and the deterioration of the neural architecture 31. These findings strongly suggest that senescence is not merely a byproduct of neurodegeneration, but a primary, causal driver of the pathology.
Pharmacological Interventions
Recognizing cellular senescence as a fundamental, modifiable biological mechanism has catalyzed the emergence of "senotherapeutics," an entirely new class of pharmacological interventions aimed at delaying, preventing, or potentially reversing neurodegenerative diseases 5051. These therapeutics are broadly categorized into senolytics, which selectively eliminate senescent cells, and senomorphics, which neutralize the SASP without inducing cell death 135052.
Senolytics for Selective Clearance
Senolytics function by exploiting a critical vulnerability of senescent cells: their strict reliance on anti-apoptotic pathways (SCAPs) to survive despite harboring immense genomic and oxidative damage. By inhibiting these survival networks, senolytics selectively trigger apoptosis in senescent cells while leaving healthy, dividing cells completely unharmed 135052.
The most extensively studied senolytic regimen is the combination of Dasatinib (a small-molecule tyrosine kinase inhibitor originally developed for leukemia) and Quercetin (a naturally occurring plant flavonoid). Together (D+Q), they inhibit a broad, overlapping spectrum of SCAP targets, including PI3K, SRC kinases, and several Bcl-2 family proteins 31350. In preclinical models of AD (such as the APP/PS1 and PS19 mice), intermittent administration of D+Q successfully penetrates the blood-brain barrier, actively clears senescent microglia and OPCs, substantially reduces neuroinflammation, and preserves both synaptic density and cognitive function 33750.
Other emerging senolytics include specific Bcl-2/Bcl-xL inhibitors, such as Navitoclax (ABT-263). While highly effective at clearing senescent cells, the clinical application of Navitoclax in the CNS is currently limited by systemic on-target toxicities, most notably severe thrombocytopenia 3. Additionally, naturally occurring phytochemicals like Fisetin are being evaluated for their robust senolytic properties and favorable safety profiles 51.
Senomorphics and Secretome Modulation
In contexts where the physical presence of senescent cells may provide necessary structural support to the tissue, or where aggressive apoptotic clearance risks further tissue damage, senomorphics offer a highly effective complementary approach 5052. Senomorphics aim to reprogram the senescent cell, shutting down the toxic SASP while maintaining overall cell viability and structural integrity 1350.
Standard senomorphics include rapamycin (a potent mTOR inhibitor) and metformin (an AMPK activator), both of which effectively downregulate the translation of pro-inflammatory cytokines like IL-6 and IL-1β 2250. JAK/STAT pathway inhibitors, such as Ruxolitinib and Baricitinib, are also utilized to attenuate microglial activation and decrease cytokine release 50.
However, newer, highly targeted senomorphics are rapidly entering development to address specific neurovascular vulnerabilities. For instance, SW033291, a small-molecule inhibitor of the 15-PGDH enzyme, has recently demonstrated remarkable efficacy in preserving the blood-brain barrier 343453. By selectively blocking 15-PGDH, SW033291 prevents SASP-mediated vascular inflammation and completely arrests neurodegeneration in mouse models of Alzheimer's disease and traumatic brain injury. This represents a major breakthrough in BBB-penetrant senomorphic design, offering a systemic method to protect the brain's immune privilege 3434.
| Therapeutic Strategy | Mechanism of Action | Primary Cellular Targets & Pathways | Key Pharmacological Agents |
|---|---|---|---|
| Senolytics | Induces apoptosis selectively in senescent cells by disabling pro-survival SCAP networks. 1350 | PI3K, Bcl-2 family proteins, SRC kinases. | Dasatinib + Quercetin (D+Q), Navitoclax (ABT-263), Fisetin. 31351 |
| Senomorphics | Suppresses the secretion of the SASP without killing the cell, halting chronic inflammation. 5052 | mTOR, NF-κB, 15-PGDH, JAK/STAT signaling. | Rapamycin, Metformin, SW033291, JAK inhibitors. 3450 |
Clinical Translation and Global Consensus
The highly compelling preclinical data surrounding senotherapeutics has recently driven the transition of these experimental drugs into human clinical trials. Two vanguard, open-label Phase 1/2 clinical trials - SToMP-AD (Senolytic Therapy to Modulate Progression of Alzheimer's Disease) and STAMINA - have provided the first critical insights into the safety, feasibility, and target engagement of senolytics in older adults with mild cognitive impairment and early-stage AD 545556.
Human Clinical Trials in Neurodegeneration
The SToMP-AD and STAMINA trials utilized an intermittent "hit-and-run" dosing strategy of oral Dasatinib and Quercetin (D+Q) over a 12-week period. Crucially, these trials confirmed the central nervous system penetrance of Dasatinib, successfully detecting the drug within patient cerebrospinal fluid (CSF) at physiologically relevant concentrations 555658. While Quercetin was not detected in the CSF, the systemic treatment proved highly tolerable across the cohort, with no serious adverse events reported 5857.
Biological activity was confirmed via significant reductions in peripheral plasma inflammatory markers and specific SASP factors, including robust decreases in TNF-α. Notably, these biochemical reductions correlated moderately with improvements in Montreal Cognitive Assessment (MoCA) scores 55. Larger, double-blind, placebo-controlled Phase 2 trials are currently underway to further evaluate long-term cognitive outcomes and structural brain preservation via advanced PET imaging 5455.
Simultaneously, alternative therapeutic approaches targeting senescence and cellular regeneration are advancing globally. In Japan, the Sumitomo/Kyoto University clinical trials are evaluating the safety and efficacy of human induced pluripotent stem (iPS) cell transplantations for Parkinson's disease, showing early signs of improved motor function 60. Furthermore, at the AD/PD 2026 International Conference, novel therapeutics, such as AriBio's AR1001 (a PDE5 inhibitor designed to reduce brain inflammation and tau levels), continue to demonstrate the broadening scope of anti-aging neurotherapeutics 58.
International Guidelines and Precision Medicine
As clinical momentum surrounding senotherapeutics accelerates, the international aging research community has recognized the critical need for methodological standardization. Collaborative global efforts, such as the World Health Organization's Clinical Consortium on Healthy Ageing (convened in late 2024), have emphasized the necessity of shifting toward integrated, precision medicine frameworks to handle the growing burden of multimorbidity in aging populations 596061.
A major output of these cross-border collaborations is the establishment of the MICSE guidelines (Minimal Information on Cellular Senescence Experimentation in vivo), published recently in Cell. These guidelines provide a universal, standardized framework for identifying and quantifying senescent cells across global laboratories, ensuring that researchers rely on a multi-marker approach rather than single, easily misinterpreted metrics 4762.
The integration of the MICSE guidelines and the findings presented at the 2025 Brain Aging Symposium at Harvard Medical School support a fundamental paradigm shift toward "precision ecosystem medicine" 5963. Within this model, neurodegenerative diseases are no longer viewed simply as localized protein aggregation events occurring in isolation. Instead, they are conceptualized as the late-stage manifestation of a systemic ecosystem collapse driven by cellular senescence, microbial imbalance, and compromised vascular barriers 63. By identifying individual senescence biomarkers and applying targeted senotherapeutics before the irreversible loss of neural architecture occurs, the medical community aims not merely to manage the symptoms of dementia, but to fundamentally alter the trajectory of human brain aging.