Cellular mechanisms of aging and longevity
Introduction to Geroscience
Geroscience is the multidisciplinary biological study of aging, focusing on the molecular, cellular, and systemic processes that drive the progressive functional decline of living organisms over time. Historically, biological aging was conceptualized as an inevitable, entropic decay - a passive accumulation of cellular damage analogous to the mechanical wear and tear of inanimate machinery. However, contemporary cellular biology has revealed aging to be a highly complex, genetically modulated, and partially malleable phenomenon 112.
Chronological age represents the primary risk factor for the vast majority of non-communicable human pathologies, including cardiovascular disease, neurodegeneration, metabolic syndrome, and cancer 34. Rather than approaching these conditions as isolated clinical entities with distinct etiologies, the geroscience hypothesis posits that targeting the fundamental mechanisms of aging at the cellular level can simultaneously delay the onset of multiple age-related conditions, thereby extending the human healthspan 567. By understanding the molecular drivers of senescence, genetic instability, and metabolic dysregulation, researchers are developing interventions that manipulate the biological clocks governing cellular lifespan.
Theoretical Frameworks of Aging
The fundamental evolutionary and biological mechanisms explaining why organisms age have generated substantial scientific debate. The field is broadly divided into damage-accumulation (stochastic) theories and programmatic (adaptive or quasi-programmed) theories. This dichotomy, which traces its conceptual roots to evolutionary biologist August Weismann in the late 19th century, remains a central axis of discussion in modern longevity research 89.
Damage Accumulation and Evolutionary Theories
Damage-based theories postulate that aging results from the stochastic and cumulative degradation of biological macromolecules - such as DNA, proteins, and lipids - over time, a process that eventually overwhelms endogenous cellular maintenance and repair mechanisms 6810. From an evolutionary biology perspective, this degradation is frequently contextualized within the framework of the "selection shadow." Because natural selection acts most strongly on genetic traits that promote early-life survival and reproductive success, the evolutionary pressure to maintain somatic integrity drops precipitously after an organism's reproductive peak, allowing damage to accumulate unhindered 11.
Classical evolutionary models heavily support this damage-centric view. The Mutation Accumulation Theory suggests that late-acting deleterious genetic mutations accumulate within populations because they exert their negative phenotypic effects only after reproduction, thereby escaping the purging force of natural selection 11. Relatedly, the Antagonistic Pleiotropy theory argues that certain genes are selected for because they offer strong fitness benefits during early development, even if they exert detrimental, pro-aging effects later in life 81112. A primary example is cellular senescence, which acts as a robust tumor-suppressor mechanism in youth but drives severe tissue degradation in old age 13. Furthermore, the Disposable Soma Theory posits a physiological trade-off in resource allocation. Because environmental resources are scarce, organisms must partition energy between reproduction and somatic maintenance. Natural selection favors heavy investment in reproduction, leaving somatic maintenance - such as DNA repair and antioxidant defense - strictly sufficient to ensure reproductive age, but mathematically inadequate to prevent long-term degradation 811.
Recent computational analyses of biological clocks provide empirical support for stochastic models. Studies utilizing large multi-omic datasets demonstrate that the accumulation of purely stochastic variations in DNA methylation and transcriptomic expression is sufficient to build highly accurate predictive aging clocks. This suggests that the precise progression of aging phenotypes can emerge directly from statistical entropy and the accumulation of molecular noise, without requiring a deliberate genetic program designed to cause death 814.
Programmed and Quasi-Programmed Theories
Programmatic aging theories, conversely, suggest that senescence is a genetically encoded biological trajectory. Proponents argue that the vast diversity of maximum lifespans across closely related species, and the existence of single-gene mutations that can drastically extend lifespan in model organisms like Caenorhabditis elegans or Drosophila, imply a level of regulatory control that is inherently incompatible with purely random wear and tear 11815. Some programmatic models, such as the Pathogen Control Hypothesis, theorize that aging evolved as an adaptive, population-level mechanism to limit the transmission of chronic infectious diseases by periodically turning over the host population 17.
However, the concept of a deliberate, adaptive "suicide mechanism" in higher mammals is highly contested, as it offers no direct reproductive advantage to the individual, raising complex issues regarding group selection 112. To reconcile this evolutionary conflict, researchers introduced the "hyperfunction" or "quasi-programmed" theory. This model frames aging not as a passive breakdown, but as the active, futile continuation of developmental and growth programs into later life 11. For example, the mechanistic target of rapamycin (mTOR) signaling pathway is absolutely essential for cellular growth, protein synthesis, and organismal development. However, when this pathway continues to run unchecked in post-mitotic adulthood, it drives cellular hypertrophy, replicative senescence, and severe age-related pathology 511. In this view, aging is a programmatic run-on rather than a purposeful death sequence.
Informational and Cybernetic Decay Models
Recent theoretical work seeks to synthesize these two opposing paradigms by viewing biology through the lens of information processing. The "Cybernetic Decay" model defines biological systems as hierarchical information-processing networks, suggesting that aging is neither purely stochastic wear-and-tear nor a dedicated genetic program. Instead, it is the progressive computational drift and loss of fidelity within developmental regulatory architectures, resulting in a loss of morphostatic control 916.
Similarly, the "Double Code Hypothesis" frames aging as the inevitable physical outcome of the tension between two distinct biological inheritance systems: the relatively rigid genome and the highly plastic epigenome. As developmental epigenetic programs unfold throughout an organism's life, the precise coordination between genetic instructions and epigenetic states progressively degrades. This degradation results in the downstream accumulation of molecular damage 10. This informational perspective effectively reframes stochastic damage not as the root cause of aging, but as a secondary, downstream consequence of regulatory entropy 10.
| Theoretical Framework | Primary Driver of Aging | Evolutionary Rationale | Clinical Implication |
|---|---|---|---|
| Damage Accumulation | Stochastic molecular wear (oxidation, random mutations, mechanical stress). | Disposable Soma; Mutation Accumulation; resources are prioritized for reproduction over maintenance. | Aging is an inevitable entropic process; interventions require continual, external damage repair protocols. |
| Quasi-Programmed (Hyperfunction) | Futile run-on of developmental and growth pathways (e.g., mTOR signaling). | Antagonistic Pleiotropy; traits highly beneficial in youth are naturally selected for, despite causing pathology in old age. | Interventions must dampen or inhibit overactive metabolic and signaling pathways (e.g., mTOR inhibitors). |
| Cybernetic Decay / Informational Drift | Loss of epigenetic fidelity and coordination between genome and epigenome. | Informational drift as the plastic epigenome loses coordination with the static genome over long timescales. | Reversing aging requires reprogramming regulatory states back to youthful epigenetic attractors. |
The Hallmarks of Aging Paradigm
To organize the immense complexity of aging research into a cohesive biological model, geroscience adopted a standardized conceptual framework known as the "Hallmarks of Aging." Originally proposed by researchers in 2013, the framework identified nine common denominators of aging that fulfill three strict criteria: they must manifest during normal physiological aging, their experimental aggravation must accelerate the aging process, and their experimental amelioration must retard aging and extend healthy lifespan 3717.
By 2023, as biological understanding advanced, the framework was expanded to encompass twelve distinct hallmarks. Subsequent academic propositions in 2024 and 2025 have further advocated for an expansion to fourteen hallmarks, incorporating extracellular matrix (ECM) alterations and macro-level psychosocial factors into the biological model 171821. The hallmarks are deeply interconnected and are generally organized hierarchically into three categories based on their role in the causal chain of cellular decline: primary, antagonistic, and integrative 19202122.
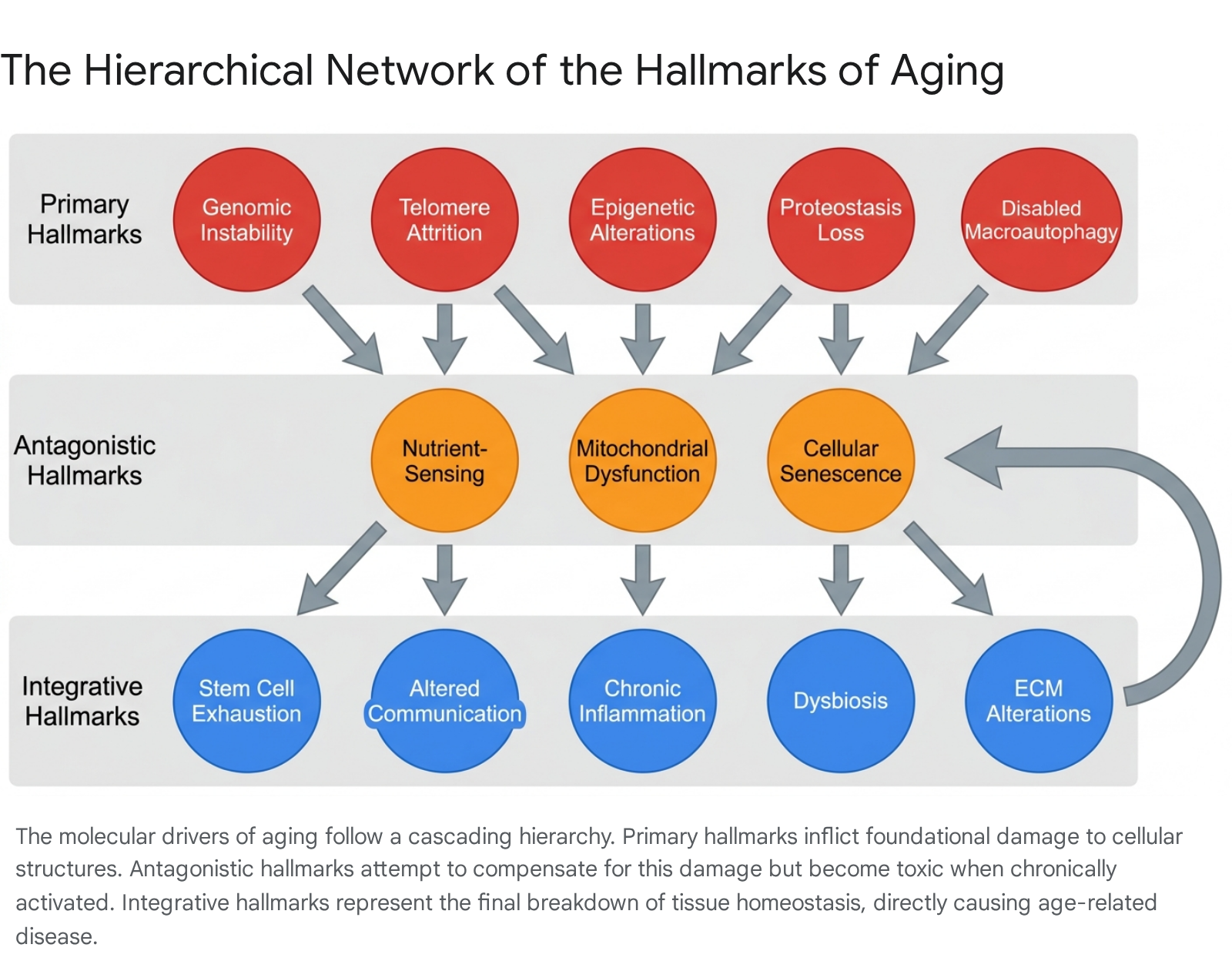
Primary Hallmarks of Aging
The primary hallmarks are unequivocally deleterious molecular events that serve as the initiating triggers of cellular damage. These phenomena accumulate relentlessly over time and form the foundational "wear and tear" of the biological system 2122.
Genomic instability represents the continuous accumulation of chromosomal alterations throughout an organism's life. Exogenous stressors (such as ultraviolet radiation and chemical mutagens) and endogenous threats (such as reactive oxygen species and DNA replication errors) constantly induce point mutations, translocations, chromosomal breaks, and copy number variations 2023. To combat this, cells rely on complex DNA repair machineries, including homologous recombination and non-homologous end joining, orchestrated by kinases like ATM and ATR. As organisms age, the efficacy of these repair pathways becomes severely impaired, resulting in a higher burden of somatic DNA lesions. This instability directly contributes to cellular senescence, widespread functional decline, and heavily elevates the risk of malignant transformation 2324.
Closely tied to genomic integrity is telomere attrition. Telomeres are tandem repetitive DNA sequences (TTAGGG) that form protective nucleoprotein caps at the ends of linear chromosomes. Because standard DNA replication enzymes cannot fully synthesize the terminal ends of linear DNA strands, telomeres progressively shorten with each cellular division. When telomeres reach a critical, minimal length, they lose their protective structure. The cell interprets this exposed chromosome end as a severe double-strand DNA break, which activates a persistent DNA damage response (DDR) and forces the cell into irreversible replicative senescence 132023.
Epigenetic alterations represent a parallel decay in informational fidelity. Age-associated changes to the epigenome include global DNA hypomethylation, localized hypermethylation at specific promoter regions, and profound alterations in post-translational histone modifications and chromatin remodeling complexes 192025. This epigenetic drift disrupts the delicate transcriptional networks required to maintain cellular function, leading to aberrant gene expression and the progressive loss of precise cellular identity, effectively causing specialized cells to forget their biological purpose 420.
At the protein level, the loss of proteostasis is a defining feature of aging cells. Proteostasis relies on a tightly regulated network of quality control mechanisms, including chaperone-mediated folding and the ubiquitin-proteasome system. With age, the efficiency of these systems declines, resulting in the toxic intracellular accumulation of misfolded, oxidized, and aggregated proteins. This breakdown is a central pathogenic mechanism in numerous age-related conditions, most notably neurodegenerative disorders such as Alzheimer's and Parkinson's disease 172120.
Originally grouped under proteostasis but classified independently in the 2023 update, disabled macroautophagy involves the specific decline in the lysosomal degradation of large cellular components and entire organelles. Healthy autophagic flux requires selective autophagy receptors like p62/SQSTM1, which bind ubiquitinated cargo and deliver it to autophagosomes via LC3 interactions. In aged cells, autophagic mechanisms stall, leading to the pathological accumulation of p62 and the failure to clear dysfunctional mitochondria and protein aggregates, severely compromising cellular resilience 131723.
Antagonistic Hallmarks of Aging
Antagonistic hallmarks are complex biological responses to the initial damage inflicted by the primary hallmarks. They exhibit pleiotropic, dose-dependent behaviors: at low or transient levels, they offer highly protective, hormetic adaptations, but when chronically activated over decades, they become major drivers of tissue pathology 192022.
Deregulated nutrient-sensing involves age-related alterations in metabolic signaling pathways, particularly the insulin/IGF-1 signaling cascade, the mechanistic target of rapamycin (mTOR), AMP-activated protein kinase (AMPK), and the sirtuin family 51726. While the robust activation of these pathways is biologically necessary for growth and development in youth, their chronic activation in older organisms becomes highly detrimental. For instance, sustained mTORC1 activation aggressively inhibits macroautophagy by phosphorylating ULK1 and ATG13, preventing the clearance of toxic cellular debris and driving cellular hyperfunction and subsequent aging 526.
Mitochondrial dysfunction emerges as the efficiency of the electron transport chain declines with age. This decay leads to diminished ATP production and significantly increased leakage of reactive oxygen species (ROS), which inflict secondary oxidative damage to cellular macromolecules. Aging mitochondria also exhibit profoundly altered dynamics, characterized by severe imbalances in mitochondrial fusion and fission (often mediated by dysregulated Drp1 and Mfn2 proteins), and a marked resistance to selective autophagic clearance (mitophagy) 172527.
Cellular senescence is perhaps the most heavily studied antagonistic hallmark. It is a state of permanent cell cycle arrest triggered by severe cellular stress, including telomere attrition, DNA damage, or oncogene activation (Oncogene-Induced Senescence). While an acute senescent response is a vital physiological mechanism that suppresses tumor formation and orchestrates localized wound healing, the chronic, systemic accumulation of senescent cells over an organism's lifespan severely disrupts tissue architecture and fuels systemic decline 51719.
Integrative Hallmarks of Aging
When the cumulative burden of damage generated by the primary hallmarks, combined with the toxic consequences of chronic antagonistic responses, overwhelms the organism's homeostatic resilience, the integrative hallmarks emerge. These systemic failures directly translate into the overt clinical phenotypes associated with aging 1921.
Stem cell exhaustion represents the progressive depletion and functional decline of tissue-resident adult stem cells. As these progenitor pools succumb to senescence, epigenetic drift, and DNA damage, organs lose their regenerative capacity, leading to phenomena such as reduced hematopoiesis, impaired muscle regeneration (sarcopenia), and diminished intestinal epithelial turnover 131720.
Concurrently, there is widespread altered intercellular communication. This involves the breakdown of endocrine, neuroendocrine, and localized paracrine signaling networks. Hormonal profiles shift dramatically with age, and the mechanical and chemical signaling required for tissue coordination becomes erratic, further isolating cells and inhibiting coordinated tissue repair 131728.
This breakdown in communication is heavily driven by chronic inflammation, commonly termed "inflammaging." Inflammaging is a pervasive, low-grade, sterile inflammatory state occurring in the absence of overt infection. It is primarily driven by the accumulation of senescent cells that secrete a toxic cocktail of inflammatory molecules, along with widespread immune system dysregulation (immunosenescence) and the chronic presence of cellular debris. This persistent inflammatory tone damages adjacent healthy cells, inhibits stem cell function, and is a foundational driver of cardiovascular disease and frailty 171828.
Furthermore, aging organisms frequently experience dysbiosis, characterized by pathological shifts in the composition and metabolic output of host microbiomes, particularly within the gut. Dysbiosis compromises intestinal barrier integrity, allowing microbial metabolites to leak into systemic circulation, thereby elevating systemic inflammation and altering nutrient absorption 1720.
Recent literature has proposed adding extracellular matrix (ECM) alterations to the integrative hallmarks. This involves the progressive loss of ECM viscoelasticity, largely driven by the accumulation of advanced glycation end-products (AGEs) and altered collagen cross-linking. Tissue fibrosis severely impairs mitochondrial homeostasis, disrupts intercellular signaling, and degrades the physical niches required for stem cell survival 1821. Finally, psychosocial isolation and mental illness have been proposed as macro-level hallmarks, acknowledging that severe psychosocial stress and psychiatric conditions trigger physiological cascades - such as hyperactivation of the sympathetic nervous system and the pro-inflammatory NF-κB pathway - that significantly accelerate biological aging at the cellular level 18.
Limitations and Critiques of the Hallmarks Framework
While the hallmarks framework dominates contemporary geroscience, it has faced substantive critique within the biological community. Critics argue that the framework relies too heavily on descriptive, associative observations rather than establishing a rigid, testable causal hierarchy 242930. Some suggest that certain hallmarks may be epiphenomena rather than root biological drivers. Furthermore, theorists argue that defining aging merely as a collection of damages fails to explain why biological systems lose their regulatory coherence in the first place. In this view, the hallmarks describe the symptomology of aging rather than providing a fundamental physical or evolutionary etiology, prompting calls for more integrated, systems-level mathematical models of biological aging 931.
Telomere Biology and Maintenance Mechanisms
Telomeres are non-coding, tandem repetitive DNA sequences bound by a specialized protein complex known as shelterin. They function as protective caps at the ends of linear chromosomes, preventing the cellular DNA repair machinery from mistaking natural chromosome ends for double-strand DNA breaks 3233.
The Mechanics of Telomere Attrition
Because the conventional DNA polymerase machinery cannot fully synthesize the extreme terminal ends of linear DNA strands (the "end replication problem"), telomeres shorten by approximately 50 to 150 base pairs with every round of cellular division 3233. Progressive telomere shortening ultimately causes the un-capping of the chromosome end. This triggers a massive and sustained DNA damage response mediated by ATM and ATR kinases, halting the cell cycle through the p53/p21 and p16INK4a/Rb tumor suppressor pathways. This permanent halt in cellular division is known as replicative senescence, or the Hayflick limit 12327. In humans, accelerated leukocyte telomere attrition correlates strongly with an increased incidence of cardiovascular disease, idiopathic pulmonary fibrosis, and diminished overall lifespan 253435.
Telomerase Activation and the Oncogenic Paradox
Telomerase is a specialized ribonucleoprotein enzyme complex that can synthesize new telomeric DNA, thereby counteracting attrition and theoretically conferring cellular immortality. The complex consists primarily of a reverse transcriptase catalytic subunit (TERT) and an integral RNA template (TERC) 3336. While telomerase is highly active in embryonic stem cells and the germline, its expression is heavily repressed in the vast majority of adult somatic tissues 3236.
The therapeutic application of telomerase activators presents a significant biological paradox. Telomere shortening evolved in large, long-lived organisms as a potent tumor-suppressive mechanism designed to strictly limit the uncontrolled proliferation of cells that have acquired dangerous mutations 3436. Reactivating telomerase therefore poses an inherent oncogenic risk. Indeed, clinical oncology demonstrates that approximately 85% to 90% of all aggressive human cancers harbor specific mutations (frequently in the TERT promoter region) that reactivate telomerase, allowing malignant cells to completely bypass replicative senescence 333637.
However, emerging in vivo research provides a more nuanced understanding of this risk. Studies evaluating AAV9-mediated TERT gene therapy in mouse models have found that transient, controlled telomerase activation did not increase the incidence of cancer, even in cohorts genetically predisposed to tumor formation 3438. By rapidly elongating critically short telomeres, the gene therapy mitigates severe genomic instability, thereby suppressing the initial chromosomal aberrations and anaphase bridges that trigger malignant transformation in the first place 3739. Because AAV vectors are non-integrating, the therapeutic TERT expression dilutes out after several cell divisions, minimizing the risk of fueling runaway oncogenesis while still providing a rejuvenative cellular burst 3738.
Meta-analyses of small-molecule telomerase activators, such as TA-65 (cycloastragenol), indicate that while these compounds can induce moderate telomere elongation in human subjects, this elongation does not consistently translate into significant functional improvements in frailty or inflammatory markers. Furthermore, while short-term severe adverse events have not been observed, long-term carcinogenic safety remains a subject of ongoing clinical debate 3540.
Alternative Lengthening of Telomeres (ALT)
The complexity of telomere biology is further underscored by the 10% to 15% of human cancers that achieve cellular immortality without relying on telomerase. These tumors utilize a homologous recombination-based pathway known as Alternative Lengthening of Telomeres (ALT) 3641. The ALT mechanism is heavily associated with loss-of-function mutations in the chromatin remodeling genes ATRX and DAXX. The absence of these proteins prevents the normal deposition of histone H3 at telomeres, causing the telomeric chromatin to shift from a tightly packed heterochromatic state to a fragile, euchromatic state that is highly permissive to rampant genetic recombination 3641.
While ALT drives extreme clinical aggressiveness in mesenchymal malignancies, such as osteosarcoma and high-grade sarcomas, it exposes unique therapeutic vulnerabilities. The immense replication stress and ongoing DNA damage inherent to the ALT process render these specific tumors highly susceptible to targeted pharmacological interventions, specifically ATR and PARP inhibitors 3641.
Cellular Senescence Dynamics
Cellular senescence is a state of stable, irreversible growth arrest. While initially characterized solely as a replication limit induced by telomere shortening, modern geroscience recognizes that senescence can be triggered prematurely by a diverse array of acute cellular stressors. These include severe oxidative stress, widespread mitochondrial dysfunction, and the aberrant activation of oncogenes (Oncogene-Induced Senescence, OIS), which serves as a critical early barrier to tumorigenesis 1527.
The Senescence-Associated Secretory Phenotype (SASP)
Senescent cells do not passively expire; they remain metabolically highly active and undergo dramatic phenotypic shifts. The hallmark of this state is the senescence-associated secretory phenotype (SASP). The SASP is a complex, highly pro-inflammatory secretome comprising cytokines (such as IL-6 and IL-8), chemokines, specific growth factors, and matrix metalloproteinases 2742.
In a youthful, healthy organism, the SASP serves a transient, highly beneficial physiological role. It recruits immune cells - specifically Natural Killer (NK) cells and macrophages - to the site of damage to clear the senescent cell, thereby facilitating tissue remodeling, aiding in wound healing, and actively preventing cancer progression 519. However, as the immune system degrades with age (immunosenescence), the precise clearance of senescent cells falters. The chronic, unmitigated accumulation of senescent cells leads to a sustained, tissue-wide SASP. This chronic secretome drives systemic "inflammaging," degrades the structural extracellular matrix, induces secondary senescence in neighboring healthy cells via paracrine signaling, and paradoxically creates a highly immunosuppressive microenvironment that can eventually foster late-life tumor metastasis 172742.
Senolytic and Senomorphic Interventions
The targeted pharmacological manipulation of senescent cells represents one of the most clinically advanced and heavily funded frontiers in modern geroscience. These therapies are broadly categorized into two distinct classes: senolytics and senomorphics.
Senolytics are specifically engineered small molecules designed to selectively induce apoptosis (programmed cell death) in senescent cells. They achieve this by pharmacologically disabling the upregulated pro-survival networks, known as Senescent Cell Anti-Apoptotic Pathways (SCAPs), that senescent cells rely on to survive their own toxic internal environments 4243. The pioneering, first-generation senolytic cocktail consists of Dasatinib (a broad-spectrum tyrosine kinase inhibitor) and Quercetin (a naturally occurring plant flavonoid). Preclinical animal trials demonstrate that clearing senescent cells with these compounds can significantly improve cardiac ejection fraction, alleviate osteoarthritis symptoms, and reverse severe age-related metabolic dysfunctions 4344. However, clinical translation to human patients faces significant hurdles. Senolytics exhibit extreme cell-type specificity, meaning a compound that clears senescent fat cells may fail entirely in lung tissue. Furthermore, early human clinical trials have shown mixed efficacy regarding significant, lasting improvements in human physical function, alongside ongoing concerns regarding potential off-target toxicity in healthy, non-senescent cellular populations 2742.
Senomorphics, conversely, do not attempt to kill the senescent cell. Instead, they are designed to pharmacologically suppress the deleterious components of the SASP. By targeting central intracellular signaling nodes like the mTOR complex or the NF-κB inflammatory pathway, senomorphics (such as Rapamycin) can effectively dampen the toxic, pro-inflammatory secretome without inducing cell death 542. While this approach is theoretically less toxic than outright senolysis, senomorphics require continuous, chronic administration to maintain suppression, carrying a significant risk of inducing broader systemic immunosuppression in elderly patients 42.
| Therapeutic Modality | Primary Mechanism of Action | Primary Physiological Advantage | Key Clinical Challenge / Risk |
|---|---|---|---|
| Telomerase Activation | Elongates shortened telomeres via exogenous TERT/TERC enzyme delivery. | Mitigates genomic instability and rescues cells from replicative senescence. | Strong theoretical risk of fueling runaway oncogenesis in cells with pre-existing mutations. |
| Senolytics | Selectively induces apoptosis in senescent cells by inhibiting SCAPs. | Directly and permanently eliminates the cellular source of the inflammatory SASP. | Off-target toxicity; clinical trials show variable and inconsistent functional improvements. |
| Senomorphics | Suppresses the SASP without killing the underlying cell (e.g., mTOR inhibitors). | Lower immediate cellular toxicity; effectively mitigates systemic inflammaging. | Requires chronic administration; carries a high risk of systemic immunosuppression. |
| Epigenetic Reprogramming | Resets aged chromatin markers via Yamanaka factors (OSK). | Targets a fundamental root cause of aging, capable of true, deep cellular rejuvenation. | High risk of complete cellular dedifferentiation and rapid teratoma formation if poorly controlled. |
Epigenetic Reprogramming Technologies
Epigenetic alterations are universally recognized as a fundamental driver of the aging process. The precise DNA methylation and histone acetylation patterns that tightly control specific gene expression gradually decay over an organism's life, leading to increased cellular entropy and a profound loss of highly specialized cellular identity 2025.
In 2006, researcher Shinya Yamanaka discovered that the exogenous introduction of just four specific transcription factors - Oct4, Sox2, Klf4, and c-Myc (collectively termed OSKM) - could completely erase these accumulated epigenetic marks. This process effectively reprogrammed adult somatic cells back into an embryonic-like state, creating induced pluripotent stem cells (iPSCs) 4549. However, the continuous, unregulated in vivo application of OSKM factors in animal models is catastrophic; it results in severe cellular dysplasia and the rapid formation of massive teratomas, as the treated cells entirely lose their specialized functions and proliferate uncontrollably 4549.
Transient Reprogramming Strategies
To harness the profound rejuvenating potential of the Yamanaka factors without inducing deadly dedifferentiation or oncogenesis, biotechnology researchers developed the highly controlled concept of "partial" or "transient" epigenetic reprogramming. By expressing the factors for strictly limited durations, or by omitting the powerful, highly proliferative oncogene c-Myc entirely (utilizing only OSK), aged cells can be gently nudged backward along the epigenetic landscape 454647.
This transient exposure recruits endogenous Ten-Eleven Translocation (TET) enzymes to initiate targeted DNA demethylation. This carefully resets the biological "epigenetic clock" and restores youthful gene expression profiles without erasing the cell's fundamental biological identity 454748. Rigorous preclinical models have demonstrated that partial reprogramming can safely restore youthful biological function to aged organs, significantly improve stem cell regenerative capacity, and successfully reverse severe vision loss in murine models suffering from crushed optic nerves or induced glaucoma 464749.
Clinical Trials in Epigenetic Rejuvenation
The field of partial epigenetic reprogramming has recently crossed a major threshold, advancing from theoretical cellular biology to tangible human clinical translation. In early 2026, the United States FDA granted investigational new drug (IND) clearance for the first-ever human trials of an in vivo partial epigenetic reprogramming therapy. The trial utilizes a proprietary gene therapy construct designated ER-100 (developed by Life Biosciences), which employs a highly controllable doxycycline-inducible system to transiently deliver OSK factors directly to patient tissues 4654.
The primary target for this pioneering trial is the human eye, specifically addressing severe, age-related optic neuropathies such as glaucoma and non-arteritic anterior ischemic optic neuropathy (NAION) 464754. The ocular environment is heavily favored for first-in-human gene therapy trials due to its natural immune privilege, the necessity for extremely low viral vector doses, and its physical isolation from the broader systemic circulation. This isolation drastically reduces the systemic oncogenic risks that have historically hindered the clinical advancement of cellular reprogramming technologies 4654.
Genetic Signatures of Exceptional Longevity
While advanced laboratory interventions offer a future pathway to engineered human rejuvenation, the natural genetic architectures of exceptionally long-lived human individuals - centenarians (living 100+ years) and supercentenarians (living 110+ years) - provide a living, naturally occurring blueprint for highly resilient aging. Extreme longevity is a highly heritable, complex polygenic trait that allows these unique individuals to significantly delay, or entirely escape, the onset of severe age-related conditions, including cardiovascular disease and Alzheimer's disease 505152.
Polygenic Adaptations and Resilience
Large-scale genome-wide association studies (GWAS) analyzing international longevity cohorts have successfully identified a range of longevity-associated variants (LAVs). The most universally replicated genetic factor across almost all populations is the strict absence of the APOE ε4 allele (a major, well-established genetic risk factor for Alzheimer's disease), combined with the robust presence of protective gene variants like FOXO3A. The FOXO3A gene operates as a master regulator, heavily influencing cellular oxidative stress resistance, controlled apoptosis, and highly efficient DNA damage repair pathways 525354. Advanced polygenic risk scoring systems (such as the CentPGS score) explicitly demonstrate that these combined longevity genotypes are strongly, inversely correlated with the clinical incidence of asthma, metabolic syndrome, and age-related cognitive decline 52.
The recent, highly detailed multiomic analysis of a 117-year-old female supercentenarian provided unprecedented, counter-intuitive insight into the mechanics of exceptional aging. Strikingly, genomic sequencing revealed that she did not harbor the canonical FOXO3A longevity allele, nor did she lack biological signs of accumulated damage. Clinical blood testing exhibited mutations highly typical of clonal hematopoiesis (specifically in the TET2 and SF3B1 genes), which in normal populations heavily elevate the risk of severe cardiovascular events and leukemia 5360. Yet, her clinical phenotype remained entirely disease-free. Researchers concluded that her extreme longevity was not derived from a lack of damage, but from a highly resilient immune system, remarkably efficient metabolic processing, and unique protective variants in chromatin remodeling pathways. These factors effectively neutralized the expected downstream pathology of her somatic mutations, demonstrating that in supercentenarians, biological aging and overt disease can be entirely decoupled 53.
Admixed Populations and Novel Genetic Variants
Historically, major centenarian studies have relied heavily on genetically homogenous populations residing in Europe, Japan, and the United States. This geographic constraint created significant scientific blind spots regarding ethnic-specific genetic effects and unique evolutionary adaptations 545556. The recent, aggressive integration of highly admixed, non-Western populations into global longevity consortia has yielded immense volumes of novel genomic data.
In Brazil, the modern population is the result of a unique, 500-year history of intense genetic admixture, combining deeply rooted Indigenous, African, European, and Japanese lineages into a single genetic pool. Researchers sequencing over 160 Brazilian centenarians and supercentenarians revealed millions of previously entirely uncatalogued genomic variants. This included over 140 unique HLA alleles that were completely absent from all major global genomic databases prior to this study 5764. These novel, population-specific variants heavily regulate immune tolerance, antigen presentation, and the long-term maintenance of T-cell populations (such as unique variations in the IL7R gene). This strongly suggests that the preservation of a highly functional, naive immune repertoire deep into the 11th decade of life is a primary biological barrier against systemic aging 5657.
Similarly, deep analyses of Italian centenarian cohorts have traced extreme longevity signatures back to DNA inherited directly from ancient Western Hunter-Gatherers (WHG) who populated Europe over 10,000 years ago, while genetic databases focused on South Asian populations in India have linked exceptional longevity to unique alleles associated with slower baseline heart rates (MYH6) and decreased risks of severe osteoporosis (ESR1) 5458. These diverse findings prove definitively that longevity phenotypes are deeply shaped by unique, population-specific demographic histories, and that global genetic diversity holds the key to uncovering novel therapeutic targets for age-related disease.
| Longevity Cohort / Demographic | Key Genetic Variants Identified | Primary Biological Mechanism Affected | Clinical Implication / Phenotype |
|---|---|---|---|
| Global / Pan-Ethnic Consensus | Absence of APOE ε4; Presence of FOXO3A | Oxidative stress resistance; DNA repair; lipid metabolism. | Universal delay in Alzheimer's onset and cardiovascular disease. |
| Admixed Brazilian Cohorts | Novel HLA alleles; IL7R variations | Antigen presentation; T-cell maintenance; immune tolerance. | Exceptional immune resilience; preservation of a naive T-cell pool past 100 years. |
| Indian Subcontinent Cohorts | MYH6, ESR1, RIMS1-KCNQ5 | Cardiac muscle function; estrogen receptor signaling; neurological health. | Slower heart rate profiles; reduced osteoporosis risk; lower anxiety/neuroticism markers. |
| Ancient European Ancestry | High proportion of Western Hunter-Gatherer (WHG) DNA | Historical evolutionary adaptations to pre-agrarian environments. | 38% higher statistical chance of reaching 100 years per standard deviation of WHG ancestry. |
Conclusion
The science of cellular longevity has transitioned dramatically from observational theories of inevitable, entropic decline to a highly precise, manipulable domain of molecular engineering. While classical evolutionary theories accurately characterize aging as the stochastic accumulation of unrepaired damage governed by harsh resource trade-offs, modern cybernetic and programmatic theories provide a deeper understanding, highlighting the decay of epigenetic information and the loss of morphostatic regulatory control.
The expanding Hallmarks of Aging framework provides a critical structural map of this highly complex process, successfully identifying primary molecular triggers, antagonistic cellular compensations, and integrative systemic failures. Although the framework is critically recognized by evolutionary biologists for being more associative than purely causal, it remains the foundational scaffolding for therapeutic translation. The current clinical landscape is advancing at an unprecedented pace, utilizing first-generation senolytics to clear toxic cellular debris, developing highly specific targeted therapies for unique telomere maintenance mechanisms, and initiating pioneering, FDA-cleared human trials for partial epigenetic reprogramming to actively reset cellular age. Augmented by the diverse genetic secrets recently uncovered in global supercentenarian cohorts, these cellular interventions robustly validate the core geroscience hypothesis: aging is a fundamentally malleable biological process, and the precise pharmacological modulation of its core cellular pathways holds the unprecedented potential to profoundly extend human healthspan.