Cellular effects of acute sleep deprivation on the brain
Introduction
Sleep is a universal biological imperative conserved across animal phyla, yet the precise cellular and molecular functions that necessitate a daily, reversible disconnection from the environment remain a subject of intensive scientific investigation. Historically, the consequences of sleep deprivation were quantified primarily through behavioral and cognitive metrics, such as lapses in vigilant attention, working memory deficits, and mood dysregulation. However, recent advances in neuroimaging, quantitative proteomics, and transcriptomics have shifted the analytical focus toward the microscopic environment of the brain.
The deprivation of sleep for a single 24-hour cycle - commonly referred to as acute sleep deprivation - acts as a profound physiological stressor. Rather than a mere absence of rest, prolonged wakefulness fundamentally alters the brain's microenvironment and cytoarchitecture. At the cellular level, acute sleep deprivation triggers an intricate cascade of pathological events: the metabolic saturation of synaptic networks, the suspension of macroscopic fluid clearance mechanisms, the initiation of neuroinflammatory cascades by resident immune cells, and the accumulation of oxidative damage within mitochondria and white matter tracts. By examining the brain after one night of sleep loss, researchers can isolate the specific homeostatic functions that sleep normally provides, revealing a highly coordinated interplay between neurons, glial cells, and the neurovascular interface.
The Synaptic Homeostasis Hypothesis
A primary conceptual framework for understanding the neurological necessity of sleep is the Synaptic Homeostasis Hypothesis (SHY), initially proposed by Tononi and Cirelli 1234. The hypothesis posits that sleep is the inescapable biological price the brain pays for waking plasticity, functioning as an essential regulator of global synaptic weight.
Wakefulness as a State of Synaptic Potentiation
During wakefulness, the brain constantly interacts with the environment, encoding new information and learning statistical regularities. This experience-dependent plasticity relies heavily on long-term potentiation (LTP), a process that strengthens the synaptic connections between active neurons 25. At the molecular level, this strengthening involves the insertion of additional AMPA and metabotropic glutamate receptors into the post-synaptic density, an increase in the physical size of dendritic spines, and a higher frequency and amplitude of miniature excitatory postsynaptic currents (mEPSCs) 12.
While essential for learning and memory acquisition, this continuous net increase in synaptic strength across the cortex is biologically unsustainable. Potentiated synapses require exponentially more energy to maintain resting membrane potentials and to synthesize, transport, and release neurotransmitters. Furthermore, continuous potentiation progressively reduces the signal-to-noise ratio within neural circuits and eventually saturates the available physical and metabolic space for further plasticity, resulting in a state where no new learning can efficiently occur 136.
The Failure of Synaptic Downscaling During Sleep Deprivation
Under normal physiological conditions, non-rapid eye movement (NREM) sleep provides the mechanism to counteract waking potentiation. NREM sleep is characterized by slow-wave activity (SWA, 0.5 - 4.5 Hz) in the electroencephalogram, driven by cortical neurons oscillating synchronously between depolarized and hyperpolarized states 24. The Synaptic Homeostasis Hypothesis dictates that this spontaneous, low-frequency activity induces global synaptic downscaling or "renormalization." Synapses are proportionally weakened across the brain, preserving the relative strength of newly formed memory circuits while eliminating the weakest synapses that represent environmental noise 57.
When an individual undergoes acute sleep deprivation, this critical down-selection process is bypassed. A single night of sleep loss results in the sustained elevation of net synaptic strength. The brain remains in a state of high metabolic demand, characterized by elevated levels of extrasynaptic glutamate and increased mean firing rates in cortical neurons 27. Genomic studies support this framework, demonstrating that 24 hours of sleep deprivation significantly alters the expression of plasticity-related genes. For instance, prolonged wakefulness induces a marked increase in the expression of Homer1a 15. The Homer1a gene locus is implicated in the homeostatic regulation of NREM slow-wave activity; its elevated expression during sleep deprivation serves as a molecular marker of mounting homeostatic "sleep pressure" and the urgent need for the brain to initiate downscaling 1.
Methodological Divergence in Synaptic Plasticity Research
It is critical to note that the effects of acute sleep deprivation on synaptic plasticity can vary significantly depending on the experimental paradigm and the specific brain region examined. While SHY proposes a global scaling mechanism, recent empirical data reveals targeted, localized vulnerabilities.
Studies utilizing different methods of sleep deprivation in rodent models have yielded somewhat divergent findings regarding dendritic spine density 8910. When animals are kept awake using novelty exposure - a method that introduces new environmental stimuli - researchers have observed an unexpected increase in dendritic spine density in specific regions, such as layer III pyramidal neurons of the prefrontal cortex in aged subjects, while simultaneously observing decreases in the CA1 region of the hippocampus 910. Conversely, sleep deprivation achieved through "gentle handling" (which minimizes novel arousal) consistently causes a net decrease in synaptic strength and a reduction in dendritic spine numbers in both the neocortex and the hippocampus 8. These variations indicate that while the overarching principle of synaptic saturation holds true, the exact physical manifestation of acute sleep loss on synaptic architecture is highly modulated by the sensory experiences occurring during the extended wakeful period.
Microglial Dynamics and Neuroinflammatory Cascades
While neurons experience the metabolic strain of saturated synapses, the brain's resident immune cells undergo drastic state transitions. Microglia, which constitute the primary innate immune defense of the central nervous system, are highly sensitive to the sleep-wake cycle and rapidly alter their morphology and function in response to acute sleep deprivation 111213.
Purinergic Signaling and Morphological Transitions
In a rested, healthy brain, microglia exist in a homeostatic state characterized by a small soma and highly complex, motile arborizations (processes) that constantly survey the surrounding neural parenchyma for damage or pathogens. Acute sleep deprivation reliably induces a phenomenon known as microglial "de-ramification" 1415. During a 24-hour period of forced wakefulness, microglial processes retract, thicken, and lose their continuous scanning motility, while the cell body enlarges - a distinct morphological shift indicative of immune activation 1415.
This activation is largely driven by purinergic signaling. Prolonged neuronal firing during extended wakefulness leads to the accumulation of extracellular adenosine triphosphate (ATP) 1116. This extracellular ATP acts as a danger-associated molecular pattern (DAMP), binding to specific purinergic receptors on the microglial surface, notably the P2X7 and P2X4 receptors 1116. The activation of the P2X4 receptor, in particular, has been closely linked to the release of inflammatory cytokines and the onset of hyperalgesia (increased pain sensitivity) following acute sleep deprivation 16.
The Cytokine Storm and the M1 Phenotype
Following purinergic activation, microglia shift toward a pro-inflammatory "M1" phenotype. This state transition is mediated by canonical inflammatory cascades, including the Nuclear Factor kappa B (NF-κB) and Mitogen-activated protein kinase (MAPK) pathways 1117. Once activated, microglia release a barrage of pro-inflammatory cytokines into the central nervous system, most notably Tumor Necrosis Factor-alpha (TNF-α), Interleukin-1 beta (IL-1β), and Interleukin-6 (IL-6) 111215.
The regulatory mechanisms that normally keep this inflammation in check also fail during sleep loss. Recent cellular analyses have identified topoisomerase 1 (TOP1) as a key molecule that restrains IL-6 secretion in microglia. During acute sleep deprivation, this restraint diminishes, contributing to unchecked inflammatory signaling 15. Concurrently, sleep deprivation downregulates vital anti-inflammatory pathways. The expression of the protective α7 nicotinic acetylcholine receptor (α7-nAChR), which normally suppresses inflammation and oxidative stress via the PI3K/AKT/GSK-3β pathway, is significantly reduced 1218. Furthermore, dopamine receptor D2 (DRD2) expression on microglia decreases, which weakens dopaminergic inhibitory signaling and allows microglia to release excessive amounts of glutamate, contributing to localized excitotoxicity and neuronal death 18.
The consequences of this cytokine release are twofold. First, high levels of TNF-α and IL-1β directly impair synaptic function, further degrading cognitive performance and vigilant attention 15. Second, these cytokines can compromise the integrity of the blood-brain barrier and leak into the systemic circulation, explaining why acute sleep deprivation is often accompanied by a rapid rise in peripheral inflammatory markers in the blood 12.
Aberrant Synaptic Pruning by Glial Cells
The interaction between microglia and synapses during sleep deprivation presents a complex biological paradox. While the Synaptic Homeostasis Hypothesis models prolonged wakefulness as leading to a net increase in overall synaptic strength across the cortex, advanced imaging studies reveal that microglia actually escalate their phagocytic activity during acute sleep deprivation 14.
Using intravital two-photon imaging, researchers have observed that during 24 hours of acute sleep deprivation, microglial expression of CD68 - a lysosome-associated membrane protein indicative of active phagocytosis - increases significantly, particularly in the CA1 region of the hippocampus 914. These activated microglia target and engulf specific dendritic spines, leading to the accelerated elimination of synaptic connections 1418.
This microglial elimination of dendritic spines during sleep deprivation is not the physiological, energy-saving "downscaling" described by SHY. Instead, it is an aberrant, stress-induced pruning process driven by neuroinflammation and metabolic dysregulation 1518. Rather than globally renormalizing synaptic weights to improve the signal-to-noise ratio, sleep-deprived microglia destructively prune active neural circuits. Evidence suggests that normal microglial function during recovery sleep is actually required to protect newly formed memory synapses from being erroneously downscaled; when microglia are experimentally ablated, animals lose these fear-conditioning memories after a period of sleep deprivation 13. Thus, acute sleep deprivation effectively shifts microglia from synapse protectors into agents of inflammatory synaptic degradation.
Glymphatic Clearance and Pathological Protein Accumulation
Perhaps the most mechanically profound discovery regarding the neurobiology of sleep in the last decade is the characterization of the glymphatic system - a macroscopic waste clearance network that relies nearly entirely on the sleep state to function efficiently.
Fluid Dynamics During Sleep Versus Wakefulness
The brain lacks a traditional lymphatic system within the parenchyma. Instead, it utilizes a glia-dependent perivascular network to clear metabolic waste. Cerebrospinal fluid flows into the brain along para-arterial spaces, driven by the pulsatility of the arteries 2019. It then crosses into the brain tissue via aquaporin-4 (AQP4) water channels, which are densely packed and polarized on the endfeet of astrocytes that wrap the blood vessels 19. The CSF mixes with interstitial fluid, collecting soluble waste products, and exits via para-venous routes to the meningeal lymphatic vessels and systemic circulation 2019.
This convective flow is highly state-dependent. During deep NREM (slow-wave) sleep, the brain's noradrenergic tone drops significantly 19. This neurochemical shift triggers a profound structural change: the extracellular interstitial space expands by approximately 60% 19. This massive expansion dramatically lowers tissue resistance, allowing a surge of CSF to wash through the parenchyma and clear out accumulated neurotoxic proteins, primarily amyloid-beta (Aβ) and tau 2019.
When an individual remains awake for 24 hours, the locus coeruleus continues to secrete norepinephrine, and the interstitial space remains contracted.
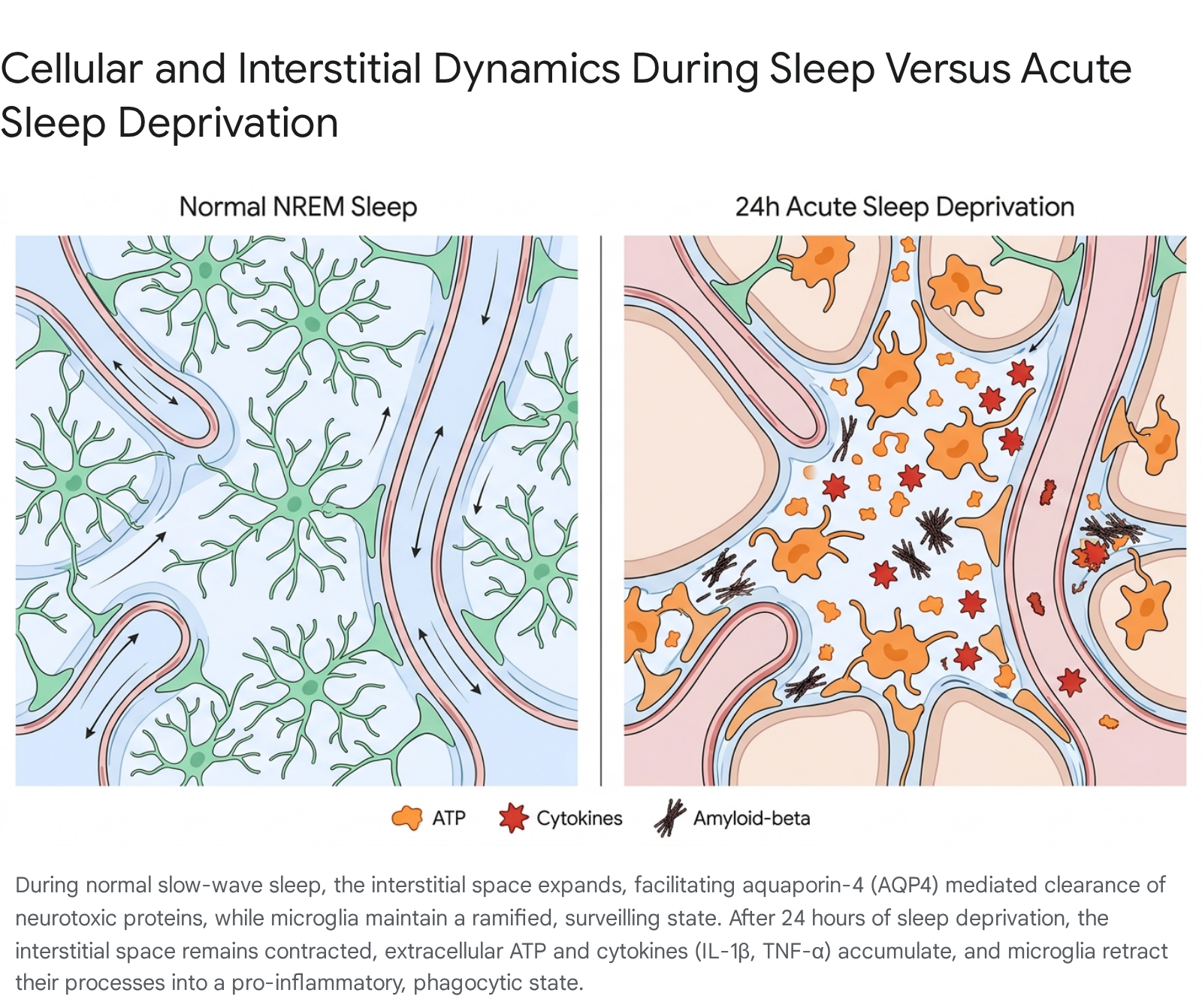
The convective flow of the glymphatic system is functionally throttled 19. Robust multi-modal imaging studies (including dynamic MRI, SPECT, and fluorescent fiber photometry) have definitively demonstrated that true clearance - the export of solutes out of the brain to the peripheral tissues - is sharply suppressed by wakefulness and enhanced only by sleep and specific anesthetics 2220.
Quantitative Biomarker Shifts Following Acute Sleep Loss
Because the glymphatic system is suspended during sleep deprivation, metabolic byproducts produced by actively firing neurons are trapped in the interstitial fluid. Human crossover trials have accurately quantified the exact burden this places on the brain. A single night of total sleep deprivation significantly alters the concentrations of Alzheimer's disease biomarkers in the cerebrospinal fluid. Because Aβ and tau are continuously released by active neurons during wakefulness, their production increases while their clearance halts 201921.
| Alzheimer's Disease Biomarker | Biological Function and Pathological Role | Quantitative Impact of 1 Night of Sleep Deprivation (CSF Levels) |
|---|---|---|
| Amyloid-beta 40 (Aβ40) | Highly abundant amyloid species; acts as a precursor to toxic plaque formation in the interstitial space. | Robust average increase of ~31.88 pg/mL compared to rested baseline 21. |
| Amyloid-beta 42 (Aβ42) | Highly fibrillogenic amyloid species; the primary structural component of Alzheimer's plaques. | Significant average increase ranging from ~34.85 to 37.32 pg/mL 21. |
| Total Tau (t-tau) | Microtubule-associated protein; elevated levels indicate axonal stress, injury, and cellular degradation. | Substantial average increase of ~37.81 pg/mL (representing a roughly 50% jump in some human cohorts) 2122. |
| Phosphorylated Tau (p-tau) | Hyperphosphorylated variant that misfolds to form destructive intracellular neurofibrillary tangles. | Modest but consistent average increase of ~1.01 pg/mL 21. |
Recent human studies have also uncovered an inverse relationship between CSF and plasma biomarkers following sleep loss. After a night of normal sleep, morning plasma levels of Aβ and tau are higher, reflecting the successful overnight export of these proteins from the brain into the systemic bloodstream 201923. Conversely, after acute sleep deprivation, morning plasma levels of these biomarkers are significantly lower, confirming that the proteins have been physically trapped behind the blood-brain barrier due to the failure of glymphatic clearance 2023.
This clearance failure is exacerbated by genetic risk factors. Apolipoprotein E ε4 (APOE4) is the most well-established genetic risk factor for Alzheimer's disease. Preclinical studies indicate that the APOE4 variant fundamentally disrupts perivascular AQP4 polarization. When APOE4 carriers undergo sleep deprivation, they suffer a compounded penalty: the genetic impairment of AQP4 channels synergizes with the wake-induced contraction of the interstitial space, leading to vastly accelerated accumulation of Aβ and tau spreading 2724.
Mitochondrial Dysfunction, Oxidative Stress, and White Matter Integrity
The metabolic toll of continuous wakefulness extends deep into the intracellular organelles. The brain consumes a disproportionate amount of the body's oxygen supply to fuel the ATP production required for continuous synaptic transmission. Extended wakefulness pushes mitochondrial respiration to its absolute limits, resulting in a breakdown of mitochondrial integrity and the accumulation of severe oxidative stress.
Reactive Oxygen Species and mtDNA Efflux
During acute sleep deprivation, the overworked electron transport chain leaks excessive reactive oxygen species (ROS), leading to the oxidative damage of local lipids, proteins, and nucleic acids 25. In rodent models, 24 hours of sleep loss results in robust brain oxidative stress, quantified by significantly elevated levels of 8-oxo-dG, a primary biomarker of DNA oxidation 25.
One of the most severe downstream consequences of this oxidative stress is physical damage to mitochondrial morphology, which eventually causes the rupture of the mitochondrial membrane. This rupture allows mitochondrial DNA (mtDNA) to efflux from the organelle into the cell's cytosol 25. In microglia, the presence of cytosolic mtDNA is recognized by the cell as a severe danger signal, triggering the activation of the cGAS-STING (cyclic GMP-AMP synthase - stimulator of interferon genes) pathway 25. The activation of cGAS-STING acts as a powerful amplifier of the neuroinflammatory response, further driving the production of pro-inflammatory cytokines and contributing heavily to the behavioral and cognitive impairments observed after sleep loss 25.
Impact on Oligodendrocytes and Myelin Thinning
The oxidative and metabolic stress of acute sleep deprivation also inflicts damage on the brain's white matter. Oligodendrocytes - the glial cells responsible for synthesizing and wrapping neuronal axons in insulating myelin - are highly vulnerable to sleep loss. Genetic analysis and electron microscopy have revealed that during acute sleep deprivation, oligodendrocytes lose their ability to efficiently metabolize and process cholesterol 26.
Cholesterol is a vital structural component of the myelin sheath. When its metabolism is impaired by sleep loss, the myelin sheath begins to physically thin 26. This reduction in insulation slows the conduction velocity of action potentials traveling along the axon, directly contributing to the "brain fog," delayed reaction times, and reduced neural synchronization across large-scale brain networks (such as thalamocortical connectivity) universally experienced after a night of sleep deprivation 2627.
Genomic Shifts and Neurotrophic Factor Decline
At the most fundamental biological level, acute sleep deprivation rapidly alters the brain's transcriptional landscape. A single night of sleep loss modifies the expression of hundreds of genes, fundamentally shifting the cell from a state of homeostatic repair to a state of triage and inflammatory defense 28.
Alongside the profound upregulation of the NF-κB inflammatory pathways and cellular stress response genes, sleep deprivation actively suppresses genes responsible for routine cellular maintenance, immune regulation, and antioxidant defense 28. A critical casualty of this transcriptional shift is the production of pleiotrophin (PTN). PTN is a secreted neurotrophic growth factor that plays a protective role in the central nervous system, particularly in maintaining the integrity of the postsynaptic membrane 2930.
Quantitative proteomics has demonstrated that 48 hours of sleep deprivation drastically reduces the secretion of PTN in the brain 29. Subsequent gene co-expression analysis revealed that the decline in PTN directly leads to reduced binding at the postsynaptic membrane in the hippocampus. This loss of neurotrophic support ultimately triggers apoptotic pathways, causing neuronal death in brain regions critical for memory consolidation 29. Based on these findings, diminished PTN levels are emerging as a reliable biological indicator of the physical neurological damage inflicted by acute sleep loss and insomnia.
Brain-Region Specificity and Cognitive Deficits
The cellular consequences of acute sleep deprivation are not distributed uniformly across the brain; specific regions display heightened vulnerability, directly mapping to the observable cognitive deficits associated with sleep loss.
The prefrontal cortex and the thalamus, which govern executive function, working memory, and vigilant attention, are particularly susceptible. Functional neuroimaging reveals that 24 hours of sleep deprivation significantly reduces metabolic activity in these regions, disrupting prefrontal-thalamic functional connectivity 27. This breakdown is responsible for the delayed reaction times, attentional lapses, and impaired risk evaluation characteristic of sleep-deprived individuals 27.
Interestingly, electroencephalogram (EEG) studies suggest that the brain attempts to functionally compensate for this regional exhaustion. While 36 hours of total sleep deprivation impairs cognitive functions in the frontal and central brain regions, researchers have observed a compensatory increase in the P3 event-related potential amplitude within the parietal regions during spatial working memory tasks 31. Despite these transient compensatory mechanisms, the overall system degrades rapidly, leading to the inescapable subjective feeling of fatigue and objective failure in sustained attention tasks 273132.
Differentiating Acute Sleep Deprivation from Chronic Restriction
While a single night of total sleep deprivation (acute SD) causes severe physiological disruptions, its cellular pathology is distinct from the effects of routinely sleeping an inadequate number of hours over a prolonged period (chronic sleep restriction). Understanding the cellular distinction between the two is vital for interpreting the long-term neurodegenerative risks associated with modern sleep habits.
| Cellular and Systemic Metric | Acute Sleep Deprivation (Single 24-48 Hour Period) | Chronic Sleep Restriction (Weeks/Months of Deficit) |
|---|---|---|
| Glymphatic Fluid Dynamics | Transiently halted. The interstitial space fails to expand, temporarily trapping proteins for the duration of the wakeful period 2019. | Persistently impaired. Long-term restriction disrupts perivascular AQP4 polarization entirely, causing chronic clearance failure 1924. |
| Protein Pathology (Aβ & Tau) | Soluble Aβ and tau spike in the interstitial fluid and CSF. This spike is largely reversible with adequate recovery sleep 2122. | Soluble proteins aggregate into insoluble fibrillar plaques (Aβ) and tangles. Facilitates the trans-synaptic spreading (seeding) of tau 2122. |
| Microglial Activation State | Acute activation (M1 phenotype), excessive localized pruning of dendritic spines, and reversible morphological de-ramification 91415. | Chronic, low-grade neuroinflammation. Persistent elevation of CD68 and eventual onset of microglial dystrophy and senescence 111733. |
| Synaptic Receptor Density | Minor or transient structural shifts; primary effects are driven by immediate ligand accumulation (e.g., extracellular ATP, glutamate) 1632. | Permanent structural remodeling, including the significant downregulation of NMDA receptor subunits (NR1, NR2A) and eNOS dysfunction 333439. |
| Cognitive and Behavioral Phenotype | Severe, immediate drops in vigilant attention, slowed motor response, and significant self-reported fatigue 2627. | Cumulative cognitive deficits. Subjects often lose the subjective perception of their own impairment despite objective neurobehavioral failure 274035. |
Both paradigms share overlapping pathological endpoints - such as elevated oxidative stress and an increased longitudinal risk of neurodegeneration - but their mechanisms differ in permanency. Chronic sleep deprivation induces irreversible structural remodeling and receptor downregulation. Conversely, the cellular damage of an acute all-nighter is heavily reliant on the efficacy of subsequent recovery sleep to clear accumulated metabolites, flush out Aβ and tau, and repair oxidized mitochondrial DNA before it triggers lasting inflammatory damage 212535.
Conclusion
The biological science of sleep deprivation reveals that sleep is not merely a passive period of inactivity, but an aggressively active and necessary cellular state. The absence of sleep for just 24 hours forces the human brain into a state of metabolic and structural crisis. Without the slow-wave oscillations required for synaptic downscaling, neural networks become saturated and metabolically exhausted. The suspension of the macroscopic glymphatic system traps neurotoxic proteins, notably amyloid-beta and tau, within the brain parenchyma. In response to mounting extracellular ATP and oxidized mitochondrial DNA, microglia abandon their homeostatic surveilling roles, morphing into pro-inflammatory agents that release destructive cytokines and aberrantly phagocytose synaptic spines. Concurrently, the failure of oligodendrocytes to metabolize cholesterol leads to the physical thinning of the myelin sheath, slowing neural processing speeds.
Ultimately, one bad night of sleep strips the brain of its essential maintenance protocols, replacing repair with inflammation and toxicity. While the brain is highly resilient and relies on subsequent recovery sleep to clear these acute deficits, the profound cellular damage incurred during a single 24-hour period of wakefulness highlights exactly why sleep disruption is increasingly recognized as a primary risk factor for cognitive decline, chronic neuroinflammation, and Alzheimer's disease.