Biological Function of Fever in the Immune System
For over 600 million years of evolutionary history, the febrile response has persisted across virtually all branches of the phylogenetic tree 1. Despite the immense metabolic costs associated with elevating core body temperature, the universal conservation of fever - from invertebrates relying on environmental thermoregulation to mammals deploying complex neuroendocrine pathways - indicates that thermal elevation is a highly intelligent, advantageous host defense mechanism. Historically misunderstood as a pathological byproduct of illness, modern thermal immunology reveals fever to be a meticulously orchestrated adaptation designed to optimize immune cell metabolism, directly inhibit pathogen replication, and enhance overall survival during infection.
Historical Conceptions of Fever
The medical and philosophical interpretation of fever has undergone significant evolution. Ancient civilizations, including the Egyptians and Mesopotamians, possessed advanced anatomical knowledge but largely feared fever, conceptualizing it either as a punitive measure enacted by evil spirits or an imminent harbinger of death 2. In classical Greek medicine, Hippocrates (5th century BC) closely linked the significance of fever to pulse rate and patient symptomatology, while Galen (2nd century AD) integrated fever into the theory of humorism, positing that health depended on the precise balance of four qualities: heat, cold, dryness, and moisture 2.
A critical paradigm shift occurred in the early tenth century when the Turkish physician Razi argued that fever was not a disease entity in itself, but rather the body's active struggle to expel disease 2. However, succeeding medical eras often reverted to viewing fever as inherently detrimental. In the eleventh century, Ibn Sina described fever as a burden on the heart that spread destructive heat through the blood, disrupting physiological functions and causing fatigue. By the seventeenth century, prevailing medical doctrine suggested that fever resulted from the harmful fermentation of chemicals in the blood 2. It is only within the last 40 to 50 years, driven by advancements in molecular biology and cellular metabolism, that intensive research has definitively established fever as a highly regulated, beneficial systemic response 2.
Evolutionary Conservation Across Taxa
The ubiquitous presence of fever across diverse biological classes underscores its evolutionary necessity. While the end goal - an elevated core temperature - is shared, the biological mechanisms utilized to achieve this state diverge fundamentally depending on an organism's metabolic capabilities.
Behavioral Fever in Ectotherms
Ectothermic organisms, which comprise invertebrates, fish, amphibians, and reptiles, lack the physiological metabolic toolkit required to endogenously generate and sustain significant heat 133. To achieve a febrile state during infection, these organisms must utilize "behavioral fever." This process involves actively altering their behavior to seek out warmer microhabitats or modifying their posture to increase heat absorption from the environment 13.
Foundational research by Matthew Kluger on the desert iguana (Dipsosaurus dorsalis) quantified the direct survival advantage of behavioral fever. When injected with the bacterial pathogen Aeromonas hydrophila and placed in environments allowing for thermal selection, the lizards actively sought heat lamps to elevate their body temperatures. Of the 13 infected iguanas allowed to mount a behavioral fever, 12 survived. Conversely, the single iguana that failed to seek heat died 45. To confirm the mechanism, a subsequent experiment administered a fever-fighting antipyretic drug to 12 infected iguanas; the five that failed to overcome the drug's effects and remained normothermic died, while the seven that successfully fought the drug to develop a fever survived 56.
This phenomenon extends to aquatic environments. In controlled studies, Nile tilapia (Oreochromis niloticus) infected with the bacterium Edwardsiella piscicida deliberately migrated from 28°C waters to 34°C waters 1. This behavioral fever was maintained for five days and correlated precisely with an enhancement in adaptive T cell immunity occurring four to six days post-infection 1.
Invertebrates also display nuanced behavioral thermoregulation. A meta-analysis of insect species demonstrated that crickets, grasshoppers, and house flies (Musca domestica) exhibit behavioral fevers of 0.5°C to 6°C following bacterial or fungal infections 3. Social insects utilize collective thermoregulation to protect their colonies. Honeybees (Apis mellifera), for instance, exhibit a "social fever" when their brood is infected with Ascosphaera apis, a heat-sensitive fungal pathogen responsible for chalkbrood disease. The fungus requires temperatures below 32°C to germinate in the larval gut. In response, adult bees aggregate over the brood comb and rapidly vibrate their flight muscles to elevate the localized nest temperature to 36°C, effectively neutralizing the pathogen 78.
Conversely, thermoregulation is highly specific to the pathogen's thermal vulnerabilities. Behavioral anapyrexia - the deliberate selection of a cooler environment - has been documented in the agile frog (Rana dalmatina). When infected with the warm-adapted ranavirus, these poikilothermic hosts actively moved to cooler zones to lower their body temperature, demonstrating that thermoregulatory behavior is an adaptive, calculated response to specific pathogenic threats rather than a universal reaction 3.
Physiological Fever in Endotherms
In warm-blooded vertebrates - mammals and birds - the febrile response is an endogenously regulated physiological process 9. The cascade initiates when the host's innate immune system detects invading pathogens. Pattern recognition receptors (PRRs), such as Toll-like receptors, identify pathogen-associated molecular patterns (PAMPs), including viral RNA or bacterial lipopolysaccharides 19.
Upon recognition, immune cells synthesize and release endogenous pyrogenic cytokines, predominantly interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α) 19. These circulating cytokines travel to the brain, specifically targeting the vascular organ of the lamina terminalis (VOLT). Because the VOLT lacks a traditional blood-brain barrier, it allows these peripheral immune signals direct access to the central nervous system 10. Here, the cytokines stimulate the synthesis of prostaglandin E2 (PGE2) via the cyclooxygenase-2 (COX-2) enzymatic pathway 1911.
PGE2 acts directly on thermoregulatory neurons situated in the ventromedial preoptic area (VMPO) of the hypothalamus, the body's primary thermostat 110. By altering the firing rates of warm-sensitive and cold-sensitive neurons, PGE2 effectively raises the organism's homeostatic temperature set-point 12.
Neurophysiological Mechanisms of Thermoregulatory Reset
The distinction between a regulated fever and unregulated thermal accumulation is paramount in clinical physiology. These states differ profoundly in their neurological control mechanisms, physiological consequences, and risk of mortality.
Hypothalamic Set-Point Adjustment
Fever is defined strictly as a regulated rise to a new, elevated set-point in the hypothalamus induced by pyrogenic cytokines 12. Once PGE2 resets the thermostat from a normative 37°C to a febrile level (e.g., 39°C), the brain perceives the body's current normative temperature as hypothermic. To bridge this gap, the hypothalamus commands the body to generate and conserve heat.
Thermogenesis is achieved through involuntary skeletal muscle contractions (shivering) and increased cellular metabolism, particularly via brown adipose tissue 1213. Heat conservation is simultaneously achieved through peripheral vasoconstriction, which shunts blood away from the cutaneous vascular beds to the body's core, thereby minimizing thermal dissipation to the environment 1213. This mechanism explains why patients developing a fever paradoxically experience chills, rigors, and cold extremities despite their core temperature actively rising 13.
Importantly, cytokine-induced fever remains a heavily regulated process. Endogenous cryogenic factors provide negative feedback, ensuring that cytokine-mediated fevers seldom exceed a ceiling of 41.1°C (106°F) 1214. This physiological upper limit protects the organism from irreversible protein denaturation and cellular necrosis 12.
The Delineation of Fever and Hyperthermia
Hyperthermia is fundamentally distinct from fever. In hyperthermic states, the hypothalamic set-point remains unaltered at roughly 37°C, but the body's physical capacity to dissipate heat is overwhelmed by extreme external environmental heat loads, prolonged exertion, or specific metabolic derangements (such as malignant hyperthermia induced by anesthetic agents or thyroid storm) 101216.
Because the set-point is normal, a hyperthermic body actively attempts to cool itself, resulting in extreme vasodilation and profuse diaphoresis (sweating). However, when heat absorption or endogenous production outpaces dissipation, the core temperature rises uncontrollably 101617. Without the regulatory ceiling present in true fever, hyperthermia can relentlessly drive body temperatures well past the critical thermal maximum of 42°C 16.
The lethality of severe hyperthermia often stems from a maladaptive cardiovascular response culminating in hyperdynamic, non-septic shock. As excessive heat triggers massive cutaneous and skeletal muscle vasodilation to dump heat, systemic vascular resistance plummets. To maintain blood pressure, the heart must sustain extreme tachycardia and high-output circulation. When this compensatory mechanism fails, the resulting myocardial strain leads to ischemia, metabolic exhaustion, contractile dysfunction, and systemic circulatory collapse 15. This paradigm accounts for the high mortality rates observed among frail and elderly populations during environmental heatwaves, such as the 2003 French heatwave, where many deaths occurred at relatively modest hyperthermic core temperatures (~39°C) due to cardiovascular failure rather than direct cellular thermal injury 15.
Table 1: Clinical Delineation of Elevated Temperature States
| Physiological Parameter | Cytokine-Mediated Fever (Pyrexia) | Hyperthermia | Hyperpyrexia |
|---|---|---|---|
| Hypothalamic Set-Point | Elevated via PGE2 signaling | Normal (~37°C) | Elevated or Normal |
| Primary Etiology | Viral/bacterial infection, inflammation | Exertional heat, prolonged exposure, adverse drug reactions | Severe systemic infection, CNS hemorrhage |
| Autonomic Response | Vasoconstriction, shivering | Vasodilation, extreme diaphoresis (sweating) | Overwhelmed, variable |
| Temperature Trajectory | Highly regulated, rarely > 41.1°C | Unregulated, can exceed 42°C | Defined strictly as ≥ 41.5°C |
| Primary Intervention | Antipyretics (to lower set-point), hydration | Rapid physical cooling, hydration | Immediate emergency physical cooling |
| Data Sources | 10121317 | 1012161715 | 101317 |
Cellular and Metabolic Enhancements
The evolutionary perseverance of the febrile response is validated by its capacity to optimize both the innate and adaptive branches of the immune system. Elevated core temperatures serve as a systemic catalyst, transforming a resting immune network into a highly aggressive, metabolically hyperactive defense apparatus.
Innate Immune System Activation
Thermal elevation significantly alters the physical and chemical environment of the host. Increased kinetic energy accelerates enzymatic reactions and enhances cellular mobility. During fever, the innate immune system experiences a marked increase in leukocyte mobility, heightened phagocytic activity by macrophages and neutrophils, and improved migration of antigen-presenting cells to the lymph nodes 1617.
Furthermore, fever exerts direct bactericidal and virucidal effects. Many obligate human pathogens are evolutionarily calibrated to thrive at exactly 37°C. When ambient temperatures rise to 39°C or 40°C, pathogen replication is severely restricted. Certain bacterial agents, such as Treponema pallidum (syphilis) and Borrelia burgdorferi (Lyme disease), lack robust heat-shock proteins and are rapidly killed or inactivated at temperatures approaching 41°C 16. Viral pathogens, including SARS-CoV-2 and influenza B, demonstrate significantly restricted entry and replication kinetics at febrile temperatures 1618. Fever also operates synergistically with pharmacological interventions; physiological hyperthermia decreases the minimal inhibitory concentrations (MICs) of numerous antibiotics, such as cephalosporins and penicillins, and reduces the structural integrity of bacterial biofilms 16.
In aquatic invertebrates, thermal immunology reveals similar mechanisms. In shrimp infected with the White Spot Syndrome Virus (WSSV), elevated water temperatures (32°C) significantly inhibited viral replication. Transcriptome analysis identified that this thermal resistance is mediated by Heat Shock Protein 70 (HSP70) interacting with the Toll4 receptor, which modulates the expression of anti-microbial peptides (AMPs), underscoring the ancient role of temperature in innate immunity 19.
Adaptive Immunity and T Cell Metabolism
Thermal stress fundamentally dictates the metabolic fate and differentiation trajectory of T cells. Immunometabolism research indicates that transient exposure to febrile temperatures (39°C) during T cell activation leads to sustained metabolic reprogramming that vastly enhances adaptive effector functions 2021.
In vitro extracellular flux analysis demonstrates that CD8+ T cells activated at 39°C exhibit heightened basal extracellular acidification rates (ECAR) and maximal glycolytic responses, indicating an anabolic shift favoring glucose metabolism 20. Concurrently, transcriptional profiling reveals a marked upregulation of nuclear-encoded mitochondrial pathways. Enhanced mitochondrial translation at febrile temperatures increases overall mitochondrial mass, facilitating superior oxidative phosphorylation (OXPHOS) 20. This metabolic surplus allows thermally stressed CD8+ T cells to produce significantly higher quantities of interferon-gamma (IFN-γ) and Granzyme B, yielding robust cytotoxic responses against tumors or virally infected cells 12021.
CD4+ helper T cell subsets exhibit similarly dramatic responses to thermal variables. Activation at 39°C increases the differentiation rate of naïve CD4+ T cells into T helper 2 (Th2) and T helper 17 (Th17) phenotypes 21. Th17 cells, critical for mucosal defense, show vastly increased production of IL-17a. Furthermore, the generation of induced regulatory T cells (iTregs) at febrile temperatures results in a population that expresses standard levels of FOXP3 but possesses a significantly reduced capacity to suppress effector T cell division 21. By blunting the suppressive action of iTregs, fever effectively removes the physiological "brakes" on the immune system, allowing for a more aggressive, uninhibited clearance of the pathogen.
The Role of Heat Shock Proteins
The stabilization of the hyperactive immune state relies heavily on the induction of heat shock proteins (HSPs). As ATP-dependent molecular chaperones, HSPs - particularly HSP70 - prevent the denaturation and aberrant aggregation of nascent proteins operating in extreme thermal environments 122.
During a severe infection, such as with Mycobacterium tuberculosis (Mtb), human monocyte-derived macrophages exposed to febrile conditions (40°C) demonstrated a significant increase in both intracellular accumulation and extracellular secretion of HSP70 compared to those kept at 37°C 22. Extracellular HSP70 serves as a highly potent immune adjuvant, linking innate pattern recognition to rapid, prolonged activation of antigen-specific CD8+ T cells 26.
Crucially, fever-induced HSP70 acts as an anti-apoptotic shield for effector cells. In the Nile tilapia model of behavioral fever, elevated temperatures triggered rapid HSP70 expression. The chaperone protein translocated to the nucleus, where it bound to extracellular regulated protein kinases (ERK1/2), facilitating their phosphorylation 1. The activation of this MAPK/ERK signaling pathway directly inhibited the cleavage of the caspase-8 and caspase-3 axis 1.
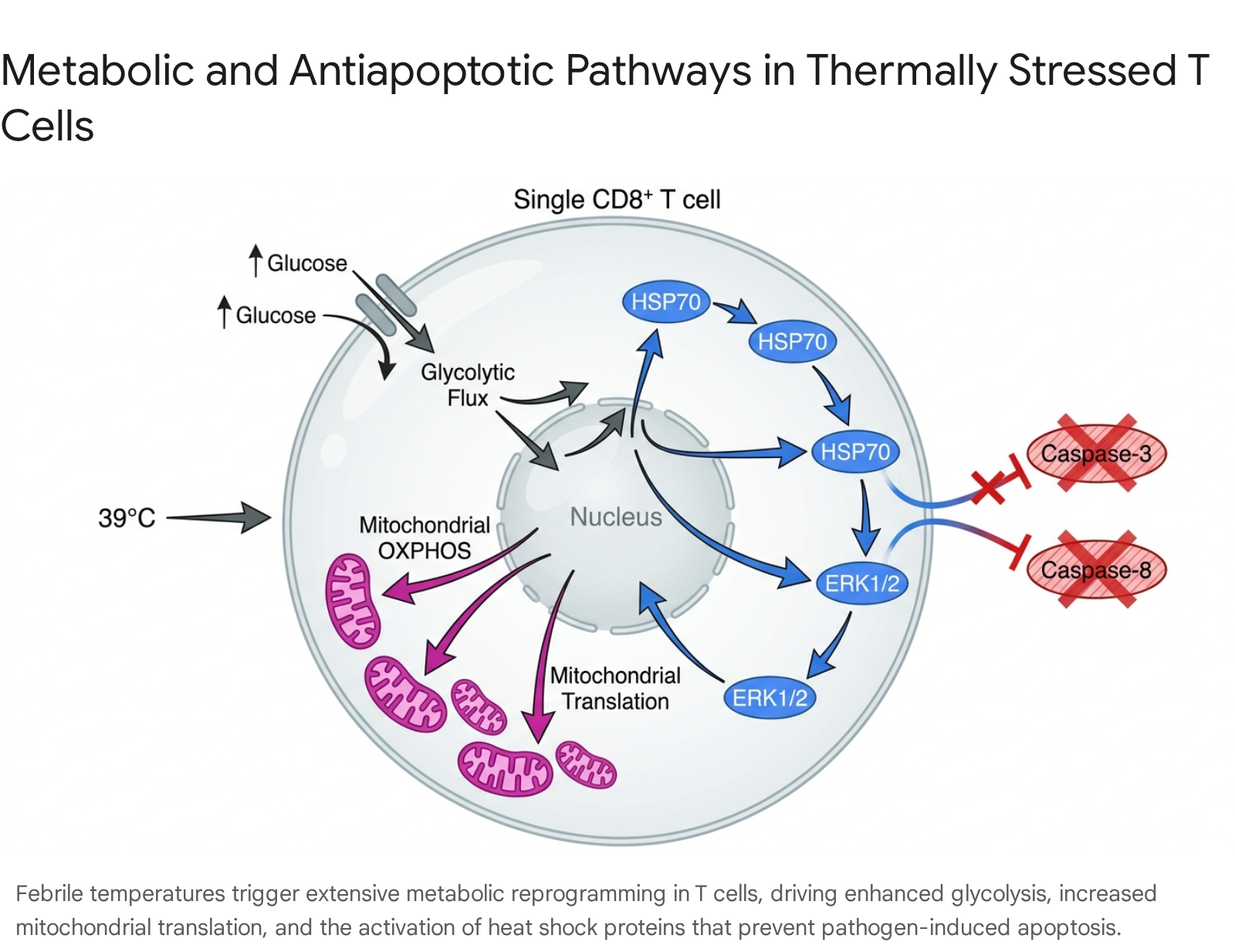
By actively blocking pathogen-induced apoptosis, the febrile response maintains a massive pool of viable, highly active T cells during the critical early days of infection, drastically reducing host mortality 1.
Limits of the Febrile Response and Metabolic Exhaustion
While fever provides unparalleled immunological benefits, it operates at a massive energetic premium. When the metabolic demands of maintaining a high core temperature surpass the host's physiological reserves, fever transitions from a protective shield to a profound systemic vulnerability.
Hyperpyrexia and Cardiovascular Strain
The metabolic cost of fever is steep: for every 1°C elevation in core body temperature, the basal metabolic rate accelerates by 10% to 13% 14. This surge demands a proportional increase in whole-body oxygen consumption and drives a 7% to 14% rise in the cerebral metabolic rate 14. To sustain this hypermetabolic state, cardiac output must increase substantially.
If regulatory mechanisms fail and fever escalates into hyperpyrexia (temperatures strictly defined as ≥ 41.5°C or 106.7°F), the organism faces an immediate medical emergency 1013. Extreme temperature elevations can precipitate acute neurologic damage, with approximately 50% of heatstroke and hyperpyrexia survivors suffering chronic cognitive or neurological deficits 10. The cardiovascular system undergoes immense strain; hyperthermic patients frequently become hypotensive despite a high cardiac output due to nitric-oxide-induced vasodilation and blood redistribution 1015. Furthermore, sustained temperatures above 40°C severely curtail blood flow to the gastrointestinal tract and can initiate disseminated intravascular coagulation (DIC), leading to multi-organ failure 1016. In these instances, the pathological damage originates not only from direct thermal injury but from severe cellular hypoxia and metabolic exhaustion 1615.
Poikilothermia and Sepsis in Vulnerable Populations
The threat of metabolic exhaustion is most pronounced in the elderly, the frail, and critically ill patients suffering from severe sepsis. With advancing age, immunosenescence and an undercurrent of chronic inflammation alter thermoregulatory networks. Older adults frequently exhibit lower basal body temperatures, meaning a reading of 37.8°C (100°F) may constitute a highly significant fever requiring clinical attention 142324.
In the context of severe sepsis or septic shock, up to 35% of patients present not with fever, but with spontaneous hypothermia (core temperatures below 35°C), a phenomenon disproportionately observed in the elderly 25. Accumulating evidence supports the "poikilothermic hypothesis" of sepsis, which suggests that this hypothermia is not a failure of thermoregulation, but an active, adaptive response to prevent metabolic exhaustion 25. By widening the interthreshold zone and mimicking the thermoregulation of cold-blooded animals (poikilothermia), the host intentionally lowers its metabolic rate, thereby reducing oxygen demand and avoiding lethal hypoxic injury to vital organs 25.
Because spontaneous hypothermia in sepsis appears to be a protective metabolic retreat, attempting to force the body back to normothermia through aggressive active rewarming can be disastrous. Clinical trials examining therapeutic hyperthermia (artificial rewarming) in hypothermic septic patients consistently demonstrated higher mortality rates 1625. Consequently, the physiological capacity of the host must dictate thermal management.
Clinical Implications of Fever Suppression
The routine pharmacological suppression of fever remains one of the most pervasive, yet scientifically contested, practices in modern medicine. Fueled by "fever phobia" - a disproportionate fear among caregivers and practitioners that fever will cause inherently irreversible brain damage or mortality - antipyretic drugs like paracetamol (acetaminophen) and non-steroidal anti-inflammatory drugs (NSAIDs) are consumed globally on a massive scale 30262728.
ICU Outcomes and General Antipyretic Efficacy
Antipyretics successfully lower the hypothalamic set-point by inhibiting the COX enzyme pathway and reducing PGE2 synthesis, offering undeniable symptomatic relief from muscle aches and malaise 30. However, lowering the temperature does not treat the underlying pathology, nor does the magnitude of a fever reliably predict the severity of the illness 1328.
In critical care, the clinical benefits of administering antipyretics to treat infection-induced fever are virtually nonexistent. A large multicenter trial involving 700 adults revealed that early administration of paracetamol to treat fever in suspected infections yielded zero improvement in ICU-free days or overall mortality at 28 and 90 days 16. Furthermore, a retrospective analysis suggested that maintaining lower temperatures via paracetamol and esophageal cooling correlated with a higher prevalence of multidrug-resistant pathogens, likely due to the suppression of the heat-dependent innate immune responses discussed previously 16. Conversely, the Fever and Antipyretic in Critically Ill Patients Evaluation (FACE) study indicated that while high fever (≥ 39.5°C) was associated with mortality in patients without sepsis, it was not an independent predictor of mortality in patients admitted with sepsis, reinforcing the nuanced, context-dependent nature of fever 14.
Antipyretics and the Prevention of Febrile Seizures
A primary medical justification for aggressively prescribing antipyretics to infants and young children has been the prevention of simple and complex febrile seizures. This clinical rationale has been thoroughly debunked by modern evidence.
A definitive 2021 Cochrane systematic review encompassing 32 trials and 4,431 pediatric patients evaluated the efficacy of various interventions in preventing the recurrence of febrile seizures 29. The synthesis of high-certainty evidence confirmed that administering prophylactic or acute antipyretic drugs (including acetaminophen, ibuprofen, and diclofenac) provides absolutely no benefit in reducing the recurrence rate of seizures during the current febrile episode or during subsequent illnesses 293031.
Clinical trials demonstrated that fever was significantly higher during episodes with seizures (39.7°C) compared to those without (38.9°C), but the administration of antipyretics failed to meaningfully alter the temperature trajectory or the neurological outcome during the critical window 30. Febrile seizures appear to be dictated by absolute peak temperatures and individual neurological susceptibility rather than the specific rate of temperature rise, rendering set-point reducers ineffective 32. The Cochrane review noted that only specific antiepileptic drugs (like intermittent diazepam or continuous phenobarbital) reduced seizure recurrence, but their use is generally discouraged due to frequent and significant adverse effects, given that simple febrile seizures themselves carry an excellent prognosis without neurological sequelae 2933.
Impact on Vaccine Immunogenicity
Perhaps the most significant, yet under-recognized, consequence of routine antipyretic use is the suppression of vaccine-induced immunity. Because fever and local inflammation are integral signs of the immune system successfully recognizing and processing a novel antigen, pharmacological suppression of this response can impede optimal immunization.
A landmark randomized clinical trial (the Prymula study) evaluated infants receiving prophylactic paracetamol to alleviate anticipated discomfort during their primary vaccination series. The study demonstrated that prophylactic paracetamol significantly reduced the antibody geometric mean concentrations (GMCs) against multiple pneumococcal serotypes included in the PCV10 and PCV13 conjugate vaccines 393435. A similar, albeit less consistent, blunting effect has been observed with ibuprofen reducing responses to pertussis and tetanus antigens 3542. The exact mechanism of this immunosuppression remains incompletely defined, but it extends beyond simple COX inhibition, likely involving disrupted leukocyte migration and impaired dendritic cell antigen presentation to naïve T cells in the early hours following vaccination 3637.
Importantly, this blunted antibody response is primarily observed following the primary vaccination with novel antigens. Upon receiving booster immunizations, the antipyretic-treated cohorts generally mount robust memory B cell responses, elevating antibody titers to accepted seroprotective levels 393435.
During the global deployment of COVID-19 mRNA vaccines, investigations analyzed whether post-vaccination analgesics compromised immunity. Surveys of individuals receiving mRNA-1273 and BNT162b2 vaccines found no evidence that using NSAIDs or acetaminophen after the onset of systemic side effects (fatigue, muscle aches, fever) reduced anti-Spike antibody levels 38. This suggests a critical temporal distinction: while prophylactic administration before immune activation is detrimental, therapeutic administration to treat established symptoms may not significantly harm vaccine efficacy 38.
Infection Outcomes and Viral Shedding
A persistent clinical question is whether reducing fever artificially prolongs the period an individual sheds live virus or extends the overall duration of the illness.
In human studies of standard respiratory infections, the data suggests antipyretics have a neutral effect. A 2023 systematic literature review and meta-analysis of 25 randomized controlled trials evaluating acute upper and lower respiratory tract infections found no statistically significant difference in illness duration or mean fever clearance time between patients receiving antipyretics and those receiving placebos 3940. Furthermore, a rigorously conducted double-blind, placebo-controlled trial in New Zealand investigated adults with PCR-confirmed influenza. Patients receiving 1 gram of paracetamol four times daily for five days showed no difference in the area under the curve for viral load, daily symptom scores, or time to clinical resolution compared to the placebo group 4142.
However, in systemic animal models of bacterial, viral, and parasitic infections, the administration of antipyretics frequently correlates with increased mortality, likely due to the loss of the metabolic enhancements to the innate and adaptive immune systems 2541. While human data on standard respiratory viruses is reassuring regarding shedding, the lack of clinical benefit from antipyretics, combined with potential adverse drug events (such as hepatotoxicity or gastrointestinal bleeding), necessitates careful consideration before defaulting to fever suppression.
Global Guidelines for Pediatric and Arboviral Fever Management
In light of the accumulated evidence detailing the protective nature of fever, major public health organizations have shifted their clinical protocols away from blanket temperature suppression, focusing instead on identifying severe pathology and managing patient discomfort.
WHO 2025 Arboviral Guidelines
The global spread of Aedes mosquitoes has placed over 5.6 billion people at risk for arboviral diseases 4344. In July 2025, the World Health Organization (WHO) released comprehensive integrated guidelines for the clinical management of Dengue, Chikungunya, Zika, and Yellow Fever, recognizing that these diseases frequently present with indistinguishable early febrile symptoms 434445.
For the management of fever and associated musculoskeletal pain in non-severe cases, the WHO strictly recommends paracetamol (acetaminophen) 4345. Crucially, the guidelines explicitly recommend against the use of any non-steroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen, regardless of disease severity. This absolute contraindication is due to the severe hemorrhagic risks associated with NSAIDs, which can catastrophically exacerbate the thrombocytopenia (low platelet count) and vascular permeability characteristics of Dengue fever 4345.
Pediatric Fever Without Source (FWS)
Managing pediatric "Fever Without Source" (FWS) remains a major diagnostic challenge. International guidelines reflect a highly stratified approach based on the age of the child and clinical biomarkers of serious bacterial infection (SBI) rather than the absolute height of the fever.
An international comparison of FWS guidelines, including those from the American Academy of Pediatrics (AAP) and the UK's National Institute for Health and Care Excellence (NICE), highlights specific consistencies 464748. For neonates younger than 28 days presenting with a fever (≥ 38.0°C or 100.4°F), guidelines universally recommend comprehensive laboratory testing (blood and urine cultures), cerebrospinal fluid analysis, and immediate empirical antibiotic treatment regardless of the infant's general appearance, due to their profound immunological immaturity and high risk of rapid sepsis 464849.
For older children (3 months to 5 years), guidelines emphasize clinical assessment tools. The NICE guidelines utilize a "Traffic Light System" (Green = low risk, Amber = intermediate risk, Red = high risk) to evaluate factors such as skin color, arousability, respiratory rate, and hydration status 5051. A high temperature alone does not mandate diagnostic escalation in this age group unless accompanied by "Red" features, such as extreme tachycardia, non-blanching rash, or unresponsiveness 5051. Furthermore, both AAP and NICE guidelines instruct healthcare providers not to use antipyretics with the sole intention of reducing body temperature, and explicitly advise against prescribing them to prevent febrile seizures. Antipyretics are indicated solely to relieve demonstrable distress and discomfort in the child 5051.
Table 2: Comparison of Major Fever Management Guidelines
| Governing Body | Target Population | Core Diagnostic Directives | Directives on Antipyretics / Interventions |
|---|---|---|---|
| WHO (2025 Arboviral) | Global populations at risk for Dengue, Zika, Chikungunya | Utilize integrated clinical features (e.g., thrombocytopenia for Dengue, arthralgia for Chikungunya) to differentiate overlapping febrile presentations. | Use paracetamol for fever/pain. NSAIDs are strictly contraindicated due to severe hemorrhagic risks. |
| NICE (UK) NG143 | Children under 5 years | Evaluate risk using the Green/Amber/Red Traffic Light System focusing on consciousness, respiratory effort, and perfusion. | Do not use antipyretics solely to reduce temperature or to prevent febrile seizures. Use only for symptomatic distress. |
| AAP (USA) | Children 0-36 months | Immediate blood/urine culture and antibiotics for neonates < 28 days. Use biomarkers (CRP, Procalcitonin) for older infants. | Treat the child's discomfort, not the thermometer reading. |
| Data Sources | 43444552 | 505153 | 46474849 |
Conclusion
The science of fever reveals a biological process of astonishing complexity and evolutionary intelligence. Far from being a harmful byproduct of infection, an elevated core temperature is an intentional, highly calibrated weapon deployed by the immune system. By resetting the hypothalamic thermostat, the body actively generates a thermal environment that drastically supercharges innate cellular mobility, optimizes the metabolic pathways necessary for robust T cell differentiation, and triggers the release of heat shock proteins that shield vital immune cells from pathogen-induced apoptosis. Simultaneously, this febrile state exploits the narrow thermal tolerances of invading pathogens, stunting viral replication and bacterial growth.
While clinical intervention remains absolutely critical when the body's regulatory mechanisms fail - such as in cases of extreme hyperpyrexia, cardiovascular strain, or sepsis-induced poikilothermia - the routine suppression of mild to moderate fever with antipyretics is increasingly unsupported by modern immunological data. Blanket temperature reduction offers no benefit in preventing febrile seizures, fails to alter the fundamental trajectory of respiratory illnesses, and carries the potential to blunt vaccine immunogenicity. By respecting fever as a vital metric of immune engagement rather than an enemy to be neutralized, modern medicine can better align with millions of years of evolutionary refinement, supporting the body's innate capacity to combat disease.