Autophagy and cellular aging
The physiological decline associated with biological aging is characterized by the progressive deterioration of cellular homeostasis. A central mechanism governing this decline is the dysregulation of autophagy, an evolutionarily conserved catabolic process responsible for the degradation and recycling of exhausted organelles, misfolded proteins, and intracellular pathogens. Derived from the Greek words for "self-eating," autophagy operates as a fundamental quality-control system that mitigates cellular stress and orchestrates metabolic adaptation. The relationship between autophagic flux and longevity is intensely complex; while robust autophagy is a recognized hallmark of cellular health and lifespan extension, its role in age-related pathologies, cellular senescence, and oncology exhibits nuanced, and often paradoxical, dynamics.
The conceptual framework of cellular self-digestion was established in the 1950s and 1960s following the discovery of the lysosome by Christian de Duve 123. For decades, the molecular architecture of the process remained opaque, largely due to the lack of genetic models and precise measurement tools 45. This barrier was broken in 1993 when Yoshinori Ohsumi utilized the budding yeast Saccharomyces cerevisiae to conduct random mutagenesis screens, identifying the first 15 autophagy-related (ATG) genes 234. Ohsumi demonstrated that when yeast lacking vacuolar degradation enzymes were subjected to starvation, autophagosomes rapidly accumulated within the vacuole, proving that a sophisticated genetic cascade orchestrated this survival mechanism 45. This seminal work, which earned the 2016 Nobel Prize in Physiology or Medicine, catalyzed modern geroscience by revealing that autophagy is not merely a passive garbage disposal system, but a highly regulated cellular defense mechanism with profound implications for human healthspan and longevity 3456.
Typology of Autophagic Pathways
Autophagy in eukaryotic cells operates through three distinct mechanisms, classified primarily by the method through which cytoplasmic cargo is delivered to the lysosome for enzymatic breakdown 789. While all three pathways converge at the lysosome, their initiation sequences, membrane dynamics, and substrate specificities diverge significantly to handle different types of cellular stress.
| Autophagic Pathway | Mechanism of Cargo Delivery | Membrane Dynamics | Substrate Selectivity and Primary Function |
|---|---|---|---|
| Macroautophagy | Cytoplasmic components are sequestered within a de novo double-membrane vesicle (the autophagosome), which subsequently transports the cargo to fuse with the lysosome 7810. | Requires extensive membrane elongation, structural modification, and vesicle nucleation coordinated by core ATG protein complexes 710. | Operates both non-selectively (bulk turnover of cytoplasm during starvation) and selectively (via receptors like p62 for the targeted removal of damaged mitochondria, termed mitophagy) 811. |
| Microautophagy | Direct engulfment of small portions of the cytoplasm and its contents into the lysosome without the intermediate formation of an autophagosome 789. | Characterized by the direct invagination (inward folding) or protrusion of the limiting lysosomal membrane to capture local cytosolic material 789. | Primarily non-selective. Plays a critical role in maintaining membrane homeostasis, regulating organelle size, and supporting cell survival under conditions of limited nitrogen 78. |
| Chaperone-Mediated Autophagy (CMA) | Direct, receptor-mediated translocation of unfolded, soluble cytosolic proteins across the lysosomal membrane 781213. | Unique among autophagic pathways in that it involves no vesicle formation or macro-membrane deformation 712. | Highly selective. Requires the recognition of a specific KFERQ-like pentapeptide motif by the Heat Shock Cognate 70 kDa protein (HSC70) and subsequent binding to the LAMP-2A receptor on the lysosome 91213. |
While microautophagy and chaperone-mediated autophagy serve vital regulatory roles, macroautophagy remains the most universally critical and extensively studied pathway in the context of global cellular aging, metabolic regulation, and disease pathogenesis 910. Therefore, unless otherwise specified, scientific discourse surrounding systemic autophagic flux generally refers to macroautophagy.
The Macroautophagy Core Gene Cascade
Macroautophagy is a dynamic, multiphase sequence that requires the precise spatial and temporal coordination of approximately 20 core ATG proteins. These proteins are traditionally categorized into distinct functional modules that drive the stages of autophagosome biogenesis: initiation, nucleation, elongation, and maturation 101415.
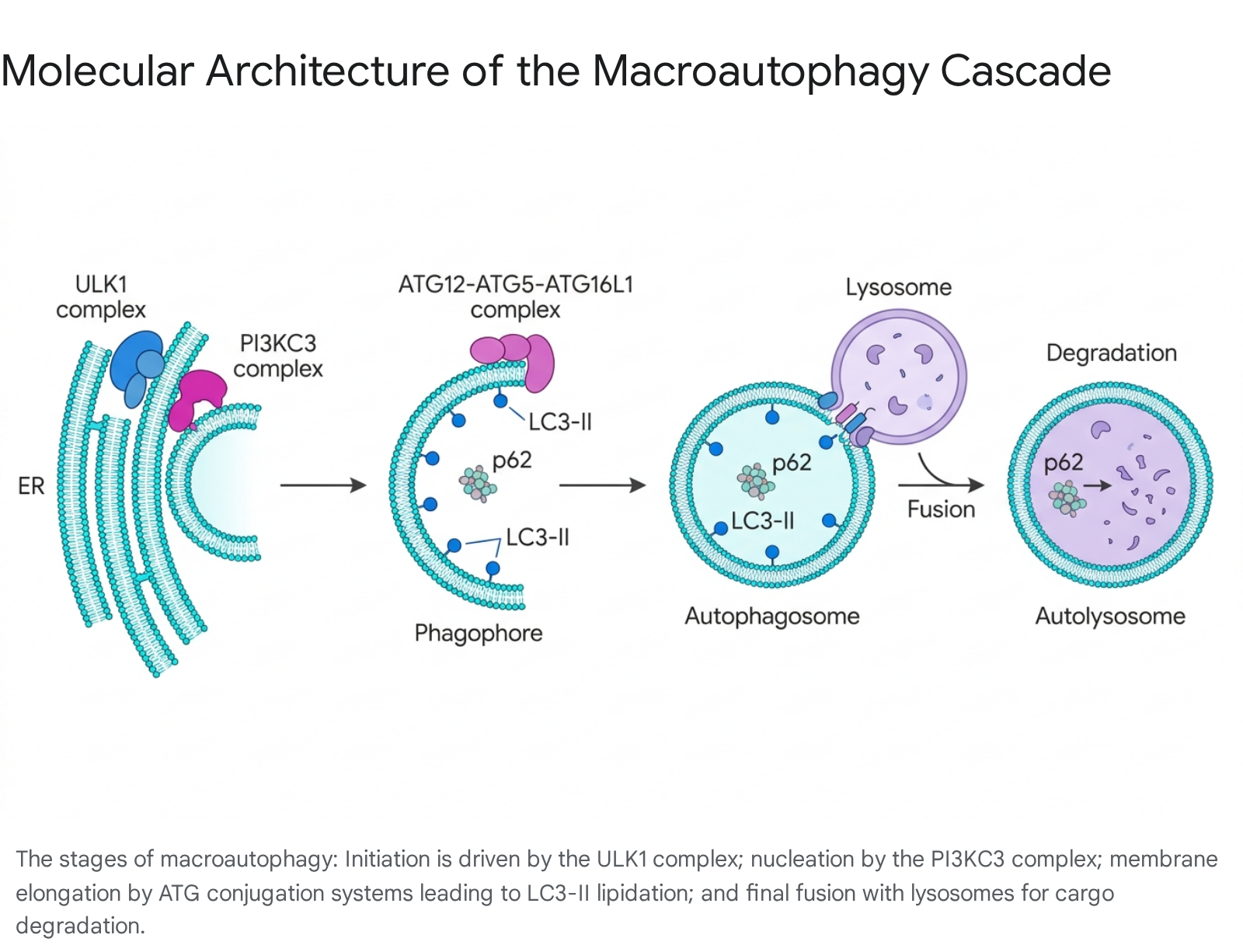
Initiation and Nucleation
The autophagic process begins with the formation of the pre-autophagosomal structure (PAS) or phagophore, predominantly originating at subdomains of the endoplasmic reticulum (ER) known as omegasomes, often at contact sites with the plasma membrane or mitochondria 111617. This initiation is governed by the unc-51-like autophagy activating kinase 1 (ULK1) complex, which includes ULK1 (or ULK2), ATG13, FIP200, and ATG101 101617. Under basal, nutrient-rich conditions, the mechanistic target of rapamycin complex 1 (mTORC1) maintains the ULK1 complex in an inactive state by phosphorylating ULK1 and ATG13 16.
When the cell experiences starvation or targeted pharmacological inhibition, mTORC1 dissociates from the complex. This release allows ULK1 to undergo activating autophosphorylation and subsequently orchestrate the nucleation phase 1516. ULK1 activation recruits the Class III phosphatidylinositol 3-kinase (PI3KC3) complex, which consists of VPS34, VPS15, Beclin1 (the mammalian homolog of yeast Atg6), and ATG14L 17. The PI3KC3 complex possesses lipid kinase activity; it phosphorylates local phosphatidylinositol to generate phosphatidylinositol-3-phosphate (PI3P). The localized accumulation of PI3P acts as a molecular beacon, recruiting effector proteins such as the WIPI (WD-repeat protein interacting with phosphoinositides) family, which tether the growing membrane and organize the structural framework for subsequent expansion 1017. In this phase, inhibitory binding between Beclin1 and the anti-apoptotic protein Bcl-2 must be disrupted, highlighting the deep intersection between autophagy and programmed cell death pathways .
Membrane Elongation and Substrate Sequestration
The expansion of the cup-shaped phagophore requires a massive influx of lipids. The transmembrane protein ATG9 (which forms homotrimers acting as a lipid scramblase) and the lipid transfer complex ATG2-WIPI synergistically deliver phospholipids from the ER and other endomembranes to construct the expanding bilayers 1015.
The physical elongation of the phagophore membrane is driven by two highly conserved ubiquitin-like conjugation systems. In the first system, the E1-like activating enzyme ATG7 and the E2-like conjugating enzyme ATG10 irreversibly conjugate the ubiquitin-like protein ATG12 to ATG5 16. The resulting ATG12-ATG5 conjugate non-covalently binds ATG16L1 to form the multimeric ATG12-ATG5-ATG16L1 complex, which acts as an E3-like ligase for the second conjugation system 1617.
Concurrently, the ubiquitin-like protein LC3 (Microtubule-associated protein 1A/1B-light chain 3) undergoes processing. Pro-LC3 is cleaved at its C-terminus by the cysteine protease ATG4 to expose a reactive glycine residue, generating the cytosolic form known as LC3-I 16. Using ATG7 as an E1 enzyme and ATG3 as an E2 enzyme, the ATG12-ATG5-ATG16L1 complex facilitates the covalent conjugation of the lipid phosphatidylethanolamine (PE) to LC3-I, producing the lipidated form, LC3-II 16.
Unlike other ATG proteins that cycle rapidly between the membrane and cytosol, LC3-II is tightly anchored to both the inner and outer surfaces of the developing autophagosome 10. LC3-II serves a dual role: it drives membrane hemifusion to seal the vesicle, and it acts as the primary docking site for autophagic cargo adaptors 1016. Receptors such as p62/SQSTM1, NBR1, and OPTN bind simultaneously to ubiquitinated targets (e.g., misfolded proteins, depolarized mitochondria, or invading bacteria) and to LC3-II, physically tethering the specific cargo to the inner membrane of the closing phagophore 101113.
Maturation, Fusion, and Degradation
Once the expanding edges of the phagophore meet and fuse, a complete, sealed double-membrane autophagosome is formed 1015. At this stage, the majority of the peripheral ATG proteins, including the ATG12-ATG5-ATG16L1 complex, dissociate from the outer membrane and return to the cytosol for reuse 10. The LC3-II localized on the outer membrane is cleaved by ATG4 and recycled, while the LC3-II trapped on the inner membrane is fated for degradation alongside the cargo 10.
The mature autophagosome relies on the cellular cytoskeleton to traffic toward the perinuclear region, where lysosomes are concentrated. Fusion between the outer membrane of the autophagosome and the lysosomal membrane is mediated by Soluble NSF Attachment Protein Receptor (SNARE) proteins. Specifically, the autophagosomal SNARE STX17 interacts with the bridging protein SNAP29 and the lysosomal SNARE VAMP8 to drive membrane fusion . The resulting hybrid organelle is termed an autolysosome 818.
Within the autolysosome, the inner autophagosomal membrane and the sequestered cargo are subjected to an extremely low pH and a battery of acidic hydrolases, including cathepsins 81219. The macromolecular cargo is catalytically dismantled into basic building blocks - amino acids, free fatty acids, and nucleotides - which are then exported back into the cytoplasm via lysosomal permeases to fuel cellular metabolism, support the synthesis of new structures, and maintain energetic homeostasis during prolonged stress 81320.
Nutrient-Sensing Pathways and Metabolic Regulation
Autophagic flux is not a static background process; it is exquisitely calibrated by an intricate network of metabolic, energetic, and stress sensors. The metabolic state of the cell dictates the equilibrium between cellular growth (anabolism) and structural recycling (catabolism). Modulations in nutritional availability seamlessly translate into biochemical signals that either halt or hyperactivate autophagosome biogenesis.
The Antagonism of mTORC1 and AMPK
The equilibrium of macroautophagy is predominantly governed by the inverse functional relationship between two master kinase complexes: mTORC1 and AMP-activated protein kinase (AMPK) 172122.
mTORC1 Signaling The mechanistic target of rapamycin complex 1 (mTORC1) functions as the primary cellular growth regulator. In the presence of abundant nutrients (particularly amino acids like leucine), growth factors, and insulin, mTORC1 is hyperactive 2324. Active mTORC1 promotes robust protein synthesis, lipid biogenesis, and cellular proliferation while simultaneously exerting a strict inhibitory grip on catabolic processes 2223. Molecularly, mTORC1 suppresses autophagy by directly phosphorylating ULK1 at Ser758 (in human cells; Ser757 in murine models) 2122. This phosphorylation event physically disrupts the interaction between ULK1 and AMPK, locking the ULK1 initiation complex in an inactive state and preventing its translocation to the pre-autophagosomal assembly site 162122.
AMPK Signaling Conversely, AMPK operates as the central metabolic checkpoint and energy sensor of the cell 2123. When the cell experiences energy stress - such as during fasting, intensive exercise, or mitochondrial dysfunction - intracellular ATP levels fall while AMP levels rise. AMP binds to the regulatory subunits of AMPK, inducing allosteric activation and facilitating its phosphorylation by upstream kinases like LKB1 2123.
Once activated, AMPK initiates a sweeping metabolic reprogramming that promotes energy-generating catabolic processes and halts energy-consuming anabolic pathways 21. AMPK stimulates autophagy through three synergistic pathways: 1. Indirect mTORC1 Inhibition: AMPK phosphorylates the tumor suppressor TSC2 (stimulating its GAP activity toward Rheb) and phosphorylates the mTORC1 subunit RAPTOR, effectively shutting down mTORC1-mediated repression 2125. 2. Direct ULK1 Activation: AMPK directly interacts with the ULK1 complex, applying activating phosphorylations at distinct sites, including Ser317, Ser555, and Ser777, driving immediate phagophore nucleation 2122. 3. Transcriptional Regulation: AMPK phosphorylates and activates transcription factors such as FOXO3, promoting their nuclear translocation where they upregulate the long-term transcription of various ATG and lysosomal genes 21.
Recent molecular insights have revealed a nuanced, secondary role for AMPK that highlights the extreme precision of cellular energy management. While AMPK powerfully initiates autophagy during moderate energy stress, research indicates that during severe or prolonged starvation, AMPK eventually phosphorylates ULK1 at an alternative site, Thr660, which paradoxically inhibits the ULK complex 2127. This feedback loop is theorized to prevent the cell from fatally exhausting its remaining, critically low ATP reserves on the energetically expensive process of autophagosome construction, effectively pausing the autophagic machinery until baseline energy levels are minimally restored 2127.
Sirtuin Signaling and Mitochondrial Quality Control
Sirtuins are an evolutionarily conserved family of NAD+-dependent histone deacetylases that function as metabolic sensors, linking cellular energy status directly to epigenetic and transcriptional regulation 2325. Because their enzymatic activity relies strictly on the availability of the coenzyme NAD+ (which rises during fasting and metabolic stress), sirtuins translate energetic depletion into cellular repair mechanisms 25.
SIRT1, the most extensively studied member, operates synergistically with AMPK. SIRT1 directly promotes global autophagic flux by deacetylating key components such as the FOXO1/3 transcription factors, multiple cytosolic ATG proteins (e.g., ATG5, ATG7, ATG8), and LC3, facilitating their assembly and functional activation 2428. Furthermore, SIRT1 activates AMPK via LKB1 deacetylation, creating a positive feedback loop that reinforces the autophagic state 24.
In the context of mitochondrial quality control - a highly specialized form of macroautophagy known as mitophagy - sirtuins localized to different cellular compartments play opposing, tightly regulated roles 25. SIRT1, SIRT3 (mitochondrial), and SIRT6 (nuclear) generally promote the selective clearance of damaged, ROS-producing mitochondria 25. Conversely, SIRT2, SIRT4, SIRT5, and SIRT7 often act as negative regulators of the process. For example, SIRT7 suppresses mitophagy by maintaining the critical ubiquitin ligase PARKIN in a deacetylated state, preventing its activation and subsequent recruitment to the outer membrane of depolarized mitochondria 25. Inhibitors of these negative regulators are currently being explored to therapeutically stimulate mitophagy in metabolic and oncological contexts 25.
Polyamine Metabolism and EIF5A Hypusination
Beyond traditional kinase cascades, emerging evidence identifies polyamine metabolism - specifically the biosynthesis of the naturally occurring polyamine spermidine - as an absolute prerequisite for autophagic induction in response to nutritional stress 262728.
Acute nutrient deprivation and the pharmacological inhibition of mTOR trigger an immediate, phylogenetically conserved surge in intracellular spermidine levels 2628. This surge constitutes the critical first step in a biochemical cascade that results in the hypusination of eukaryotic translation initiation factor 5A (EIF5A). Hypusination is a rare, highly specific post-translational modification utilizing the spermidine molecule. Activated, hypusinated EIF5A selectively facilitates the translation of the Transcription Factor EB (TFEB) 2628. TFEB serves as the master transcriptional regulator of the autophagy-lysosomal network; upon translation, it migrates to the nucleus to induce a massive coordinated upregulation of autophagic and lysosomal genes 1926.
The reliance on polyamines is profound. In models spanning yeast, nematodes, flies, and mice, the genetic knockdown or pharmacological blockade of the enzyme ornithine decarboxylase 1 (ODC1) - which produces spermidine - completely abolishes the autophagy-enhancing and lifespan-extending benefits of both caloric restriction and the drug rapamycin 262728. This indicates that spermidine is not merely an independent "caloric restriction mimetic," but rather an obligatory downstream bottleneck; without a functional polyamine pathway, the upstream suppression of mTOR fails to translate into functional autophagic flux 2628.
The Paradigm of Aging and Cellular Senescence
The traditional model of biological aging posits that basal autophagic flux universally declines across all tissues over time 293031. This generalized failure of quality control theoretically leads to the toxic accumulation of misfolded protein aggregates, dysfunctional mitochondria, and lipofuscin deposits - phenomena intrinsically linked to neurodegeneration, sarcopenia, and metabolic dysfunction 203132. However, recent multi-tissue analyses and molecular profiling have revealed a reality that challenges this simplistic narrative.
Tissue-Specific Variability and Novel ATG Functions
Contemporary research demonstrates that autophagic alterations during aging are neither uniform nor universally characterized by decline 30. While tissues such as skeletal muscle and the central nervous system frequently exhibit diminished autophagic capacity with age, other tissues demonstrate localized increases in autophagic activity, likely serving as a compensatory adaptive response to escalating cellular damage 3031.
Furthermore, researchers have uncovered that core ATG proteins possess non-canonical functions that influence cellular survival independently of traditional lysosomal degradation 2029. For instance, a 2024 study investigating neurons in the nematode C. elegans revealed that the protein ATG-16.2 mediates the formation of "exophers" - large extracellular vesicles used by stressed neurons to violently extrude massive protein aggregates when the traditional autolysosomal system is overwhelmed 29. Inhibiting early-acting autophagy genes in specific neurons, rather than late-acting ones, unexpectedly extended lifespan, suggesting that the partial manipulation of the ATG cascade yields complex survival dynamics that defy the notion that "more autophagy is always better" 29.
Cellular Senescence and the Threshold Model
The nuanced relationship between autophagy and aging is most evident in the context of cellular senescence. Senescence is a state of irreversible, permanent cell-cycle arrest accompanied by a senescence-associated secretory phenotype (SASP) 3233. Senescent cells remain metabolically active but secrete a potent cocktail of pro-inflammatory cytokines, growth factors, and proteases that disrupt local tissue homeostasis and drive systemic "inflammaging" 283233.
The interplay between autophagy and senescence is profoundly paradoxical 32. Under physiological conditions, highly functional basal autophagy acts as a primary defense mechanism, delaying the onset of senescence by mitigating oxidative stress, clearing depolarized mitochondria, and preserving genomic integrity 3233. However, once cellular damage surpasses the point of no return and senescence is established, the role of autophagy completely inverts.
Established senescent cells rely on an unusually high rate of autophagic flux to survive 32. The immense metabolic and energetic demand required to continuously synthesize and secrete SASP factors forces senescent cells to aggressively break down their own intracellular components for raw materials 32. To resolve this paradox, researchers propose a "threshold model." Below a critical threshold of genomic or oxidative damage, robust autophagy protects the cell and suppresses senescence. However, once damage exceeds this threshold, autophagy transitions into a pathological support mechanism indispensable for the maintenance and persistence of the toxic senescent state 32. Consequently, the indiscriminate, systemic hyperactivation of autophagy in advanced age could theoretically backfire by inadvertently fortifying harmful populations of senescent cells, highlighting the need for highly targeted, phase-specific therapeutic interventions 32.
The Oncological Paradox: Tumor Suppression vs. Survival
The bidirectional, context-dependent nature of autophagy observed in senescence mirrors its role in clinical oncology, where the pathway functions as a double-edged sword inextricably linked to the stage of tumor development and the tumor microenvironment 34353637.
Early-Stage Tumor Suppression
In healthy tissue and the earliest stages of oncogenesis, autophagy acts as a robust tumor suppressor 343637. By maintaining stringent protein and organelle quality control, autophagy prevents the accumulation of damaged mitochondria, thereby severely limiting the production of reactive oxygen species (ROS) that induce primary DNA mutations and genomic instability 343537. Furthermore, autophagy actively degrades potentially oncogenic protein aggregates and limits the inflammation associated with necrotic cell death 3637.
The tumor-suppressive role is clearly demonstrated in genetic models. The haploinsufficiency or complete genetic deletion of critical early-stage autophagic regulators, such as Beclin1 (ATG6), results in significantly higher rates of spontaneous malignancies in diverse tissues 3538. In early-stage pathologies, boosting autophagic clearance serves to preserve cellular integrity and halt malignant transformation 3536.
Late-Stage Tumor Promotion and Drug Resistance
Once a malignancy is firmly established, the evolutionary pressure of the tumor microenvironment forces a fundamental inversion of autophagy's role; it transitions from a protective mechanism for the host into a critical survival strategy for the cancer cell 343637.
Solid tumors rapidly outgrow their surrounding vascular supply, resulting in microenvironments characterized by severe hypoxia, nutrient deprivation, and growth factor withdrawal 343637. To survive these hostile metabolic conditions, cancer cells aggressively upregulate basal autophagy, continuously recycling bulk cytoplasm to meet the intense energetic demands of rapid proliferation 343637. Furthermore, autophagy facilitates metastasis by allowing detached tumor cells to evade anoikis - a form of programmed cell death normally triggered by the loss of extracellular matrix adhesion - enabling them to survive transit through the circulatory or lymphatic systems 363738.
Critically, autophagy represents a primary biological mechanism of acquired therapy resistance 343538. When solid tumors are exposed to the genotoxic stress of cytotoxic chemotherapy, radiation, or targeted molecular therapies, the resulting massive cellular damage triggers a rapid, adaptive autophagic response 3538. By aggressively sequestering and degrading damaged organelles and misfolded proteins before apoptotic signaling cascades can reach a terminal threshold, autophagy allows cancer cells - particularly resilient cancer stem cells (CSCs) - to evade therapy-induced cell death 353638.
Consequently, the pharmacological landscape in oncology frequently investigates the administration of late-stage autophagy inhibitors (such as chloroquine or hydroxychloroquine, which prevent lysosomal acidification and fusion) alongside standard chemotherapy regimens to re-sensitize resistant tumors 3538. However, managing the systemic toxicity of global lysosomal inhibition while specifically targeting the tumor compartment remains a formidable clinical challenge 35.
Interventions Modulating Autophagic Flux
The immense therapeutic potential of optimizing autophagic pathways has driven a global surge in research aimed at identifying behavioral, dietary, and pharmacological interventions capable of inducing the process safely and sustainably.
Dietary Interventions: Caloric Restriction vs. Intermittent Fasting
Dietary restriction is universally recognized as the most potent physiological trigger for macroautophagy. When exogenous nutrient supply is withdrawn, insulin levels drop while glucagon rises; this endocrine shift results in the suppression of mTORC1 and the hyperactivation of AMPK, initiating widespread autophagic clearance in hepatic, muscular, and neuronal tissues 21394041.
While both continuous Caloric Restriction (CR) and Intermittent Fasting (IF) activate these nutrient-sensing pathways in animal models, their comparative efficacy for overall longevity has been the subject of intense debate 3941. In humans, small-scale prospective studies suggest that IF protocols (such as dawn-to-dusk fasting or time-restricted eating) lead to an upregulation of autophagy-related genes (e.g., LAMP2, LC3B) in peripheral blood, yet these studies remain constrained by small sample sizes, brief intervention periods, and the historical difficulty of directly measuring flux in human tissues 3945.
The comparative efficacy of these diets was profoundly clarified in a landmark 2024 study conducted by researchers at The Jackson Laboratory and published in Nature 424348. Tracking nearly 1,000 genetically diverse mice across five distinct dietary regimens, researchers discovered that continuous CR exerted a significantly greater impact on lifespan extension than IF 42434844. Mice subjected to the most restrictive CR diet (consuming 60% of baseline calories) lived an average of 34 months, compared to 25 months for unrestricted controls. Conversely, mice on intermittent fasting diets experienced a more modest extension, averaging 28 months 4348.
Crucially, the study dismantled the prevailing assumption that overt metabolic improvements - such as weight loss and reduced adiposity - are inherently indicative of delayed aging. The researchers discovered the phenomenon of "genetically encoded resilience" 4844. The specific animals that achieved the most profound lifespan extensions under CR were those that managed to maintain their body weight and immune cell health despite the severe caloric deficit 424348. Conversely, animals that rapidly lost substantial body fat on restrictive diets exhibited compromised energetic capacity, failing immune systems, and died prematurely 424844. As lead researcher Gary Churchill noted, the ability to retain weight in the face of metabolic stress is the true marker of longevity, demonstrating that human responses to dietary restriction will be highly individualized based on genetic context, and that chasing weight loss through severe restriction can paradoxically shorten lifespan 424348.
Pharmacological Modulators: Rapamycin and Metformin
The practical difficulty and variable safety profile of long-term severe dietary restriction have fueled the search for "CR mimetics" - pharmacological agents capable of inducing an autophagic state without requiring severe nutrient deprivation.
Rapamycin Rapamycin (sirolimus), originally discovered in soil bacteria from Easter Island and FDA-approved as an immunosuppressant for organ transplantation, is the most direct and potent pharmacological inhibitor of mTOR 214546. It is currently the most consistently validated life-extending compound in mammalian models, regularly increasing mouse lifespan by 10% to 25% across multiple independent laboratories 4547.
Translating rapamycin's benefits to healthy humans remains an ongoing challenge. The much-anticipated PEARL trial (Participatory Evaluation of Aging with Rapamycin for Longevity), the first randomized, double-blind, placebo-controlled trial evaluating rapamycin for longevity in healthy older adults, concluded and published results in 2025 454849. Testing dosages of 5mg or 10mg delivered weekly over 48 weeks, the trial failed to meet its primary endpoint, showing no significant reduction in visceral fat 45. However, secondary outcomes demonstrated statistically significant improvements in lean tissue mass and pain reduction for women taking the 10mg dose, alongside enhanced self-reported emotional well-being 4546. Crucially, the trial confirmed that low, intermittent dosing of rapamycin over a year is generally safe and does not produce the severe immunosuppression associated with the daily high doses used in transplant medicine 45. Academic trials, such as the VIBRANT trial investigating rapamycin's ability to slow ovarian aging, continue to explore specific, tissue-level applications 45.
Metformin Metformin, the ubiquitous first-line oral medication for type 2 diabetes, acts primarily by inhibiting complex I of the mitochondrial electron transport chain. This mild inhibition slightly lowers cellular ATP levels, which triggers the activation of AMPK, thereby subsequently promoting autophagy and suppressing mTORC1 5550. Over decades of clinical use, retrospective epidemiological data has consistently revealed a startling paradox: diabetic patients managing their disease with metformin frequently outlive non-diabetic individuals who do not take the drug, while also exhibiting lower incidences of certain cancers and cardiovascular events 5550.
The geroprotective efficacy of metformin in non-diabetic populations, however, remains highly contested. Critics point to poorly designed observational studies and argue that human clinical data does not support its use as a true longevity drug 51. To resolve this definitively, the Targeting Aging with Metformin (TAME) trial is currently underway. Spanning 14 U.S. clinical research centers and enrolling over 3,000 participants aged 65 to 79, TAME is designed not to track overall mortality, but rather the delayed onset of a cluster of age-related comorbidities, including heart disease, cancer, and cognitive decline 55505253. Regardless of the ultimate statistical outcome, TAME represents a profound regulatory milestone: an unprecedented attempt to compel the FDA to recognize "aging" as a legitimate, treatable therapeutic indication, paving the way for future geroscience trials 5152.
Targeted Nutritional Interventions: Spermidine and Urolithin A
Due to the complex regulatory pathways required for generic pharmaceuticals, naturally occurring, food-derived metabolites that stimulate specific autophagic networks have emerged as a primary focus of preventative longevity medicine.
| Nutritional Compound | Primary Cellular Target | Mechanism of Action | Clinical Trial Status (2025) |
|---|---|---|---|
| Spermidine | EIF5A Hypusination / TFEB | Induces systemic macroautophagy by facilitating the translation of master autophagic and lysosomal gene networks. Serves as an obligatory downstream effector for mTOR inhibition 2628. | Recent double-blind, placebo-controlled trials utilizing high-purity spermidine (hpSPD) at 40mg/day demonstrated excellent safety and tolerability over 28 days with no adverse hematological events 545556. Multi-intervention studies (combining spermidine, exercise, and anti-inflammatories) are currently enrolling to target functional healthspan 57. |
| Urolithin A | Mitophagy | A gut microbiota-derived metabolite of ellagitannins (found in pomegranates). Selectively upregulates the degradation of defective mitochondria (mitophagy) without inducing global mTOR inhibition 585966. | The 2025 MitoImmune trial (Nature Aging) confirmed that 1,000mg/day improves immune resilience in older adults by increasing naïve CD8+ T-cells and reversing markers of immune exhaustion 6061. Prior trials demonstrated significant (~12%) improvements in muscle strength and endurance parameters 585961. |
Methodological Advancements: Measuring Flux in Humans
Historically, the most severe impediment to translating autophagic research from animal models to the clinic has been the inability to accurately and safely measure the process in living humans 39456962.
Autophagy is a dynamic process; measuring its health requires assessing "flux" - the rate at which cargo is sequestered, delivered, and ultimately degraded by the lysosome 181962. Relying on static measurements of autophagosome number or static concentrations of the LC3-II protein is profoundly misleading. An observed high level of LC3-II could indicate robust, healthy autophagic induction, or, critically, it could indicate a pathogenic blockage in lysosomal fusion where autophagosomes are accumulating because they cannot be cleared (a state associated with neurodegenerative diseases) 18196263.
To accurately measure flux, scientists must perform an assay evaluating LC3-II levels in two separate states: one at baseline, and one following the application of a lysosomal inhibitor (such as chloroquine, bafilomycin A1, or concanamycin A) 196962. The mathematical difference between the inhibitor-treated state and the baseline state represents the true, active rate of autophagic degradation 18196962.
Until recently, implementing this flux assay in human patients was impossible, as administering systemic lysosomal inhibitors to individuals is highly toxic. However, a major methodological breakthrough established and refined between 2021 and 2025 allows for the direct measurement of human autophagic flux utilizing peripheral blood mononuclear cells (PBMCs) 696465.
Instead of relying on invasive muscle or liver tissue biopsies, clinicians draw a standard sample of whole blood 6465. Crucially, the blood is not extensively processed or cultured in artificial media; the ex vivo whole blood sample is immediately treated with a lysosomal inhibitor (like chloroquine) while the cells remain suspended in the patient's own serum 196962. This vital step preserves the individual's exact systemic, nutritional, and endocrine environment (e.g., circulating insulin, circulating amino acids, and specific glucose levels), ensuring the autophagic response measured accurately reflects the patient's real-time metabolic state 196962. Following incubation, the PBMCs are isolated, and advanced flow cytometry or western blotting is used to quantify LC3B-II accumulation 6965.
This optimized PBMC blood assay finally bridges the translational gap. It provides a minimally invasive, physiologically relevant, and highly accurate biomarker to longitudinally track how dietary interventions, aging, and drugs like rapamycin actively modulate autophagic flux in individual human patients 196965.
The Global Clinical Trial Landscape
Equipped with improved biomarker assays and validated preclinical data, the clinical trial landscape for aging and autophagy interventions is undergoing rapid global expansion. The clinical trials market in the Asia-Pacific (APAC) region alone was valued at USD 12.1 billion in 2024 and is projected to grow at a compound annual growth rate (CAGR) of 8.4% through 2032 66. Nations such as Australia have positioned themselves as premier global hubs for early-phase longevity trials. Leveraging robust Research and Development tax incentives (up to 43.5%) and a highly efficient Clinical Trial Notification (CTN) scheme, regulatory authorities in Australia can approve novel, first-in-human studies in as little as four to six weeks 6667.
Simultaneously, leading academic institutions worldwide are formalizing interdisciplinary geroscience initiatives. Organizations such as the Buck Institute for Research on Aging, the European DRIVE consortium (Driving next-generation autophagy researchers towards translation), and Japan's newly established Autophagy and Anti-Aging Research Center at Nara Medical University represent a concerted international effort to operationalize these discoveries 686970. By moving the field away from purely theoretical animal gerontology and toward rigorous, placebo-controlled human efficacy trials, these consortiums are standardizing the evaluation of geroprotective compounds and setting the stage for transformative preventative medicine 686970.
Conclusion
Autophagy is a foundational pillar of cellular quality control, serving as an essential defense mechanism against the molecular and organellar deterioration that drives the biology of aging. However, the translation of this mechanism into clinical reality is obstructed by immense biological complexity. The autophagic pathway is not unilaterally beneficial; while robust macroautophagy in youthful, healthy tissue suppresses genomic damage, mitigates oxidative stress, and maintains metabolic homeostasis, its role is frequently hijacked in advanced pathologies. In the context of established oncology and locked-in cellular senescence, robust autophagic flux becomes a survival mechanism for diseased cells, driving therapy resistance and systemic inflammaging.
Therefore, the future of geroscience relies on precision modulation rather than systemic hyperactivation. Discoveries spanning from the differential regulation of mTOR and AMPK, to the absolute requirement of polyamines like spermidine, and the revelation that the lifespan benefits of caloric restriction are deeply intertwined with genetic resilience, underscore the necessity of targeted approaches. With the recent advent of ex vivo blood assays capable of measuring true, dynamic autophagic flux in humans, coupled with the rollout of rigorous, multi-center clinical trials evaluating compounds like metformin, rapamycin, and urolithin A, the field is rapidly crossing the translational gap. Determining how to safely and sustainably calibrate autophagic activity in a tissue-specific, age-appropriate manner remains the ultimate frontier in the pursuit of expanding human healthspan.