Artificial intelligence and machine learning in materials science
Introduction
The discovery, synthesis, and optimization of advanced materials form the foundation of modern technological progress, driving innovations across energy storage, semiconductor manufacturing, heterogeneous catalysis, and environmental remediation. Historically, the identification of novel materials has relied heavily on Edisonian trial-and-error experimentation, physical intuition, and serendipity. These conventional pipelines are constrained by their intensive resource requirements and inherently slow developmental cycles. In recent decades, computational approaches rooted in quantum mechanics, particularly Density Functional Theory (DFT), provided a systematic method for high-throughput virtual screening. DFT allowed researchers to compute the electronic structure and ground-state energies of theoretical compounds before attempting physical synthesis. However, because the time complexity of solving the Schrödinger equation via DFT scales steeply - typically $O(N^3)$ to $O(N^7)$, where $N$ represents the number of atoms - first-principles calculations are functionally limited to small unit cells and extremely short temporal simulations 12.
The introduction of artificial intelligence (AI) and machine learning (ML) has catalyzed a fundamental paradigm shift, often referred to as the "fifth paradigm" of scientific discovery 1. By decoupling predictive accuracy from the immense computational expense of first-principles mathematics, AI algorithms can navigate a practically infinite chemical space. To contextualize this scale, the theoretical space of stable inorganic materials exceeds $10^{10}$ combinations for quaternary compounds alone, while the space for drug-like organic molecules is estimated at $10^{60}$ 3.
The current landscape of AI in materials science is defined by three interconnected advancements. First, generative AI models propose novel, thermodynamically stable crystalline structures tailored to specific functional constraints. Second, universal machine learning interatomic potentials (uMLIPs) emulate the complex physical interactions of these atoms, enabling large-scale molecular dynamics simulations at a fraction of the cost of DFT. Finally, autonomous robotic platforms - commonly known as self-driving laboratories - integrate these predictive models with high-throughput hardware to physically synthesize and characterize the proposed materials, creating a closed-loop discovery ecosystem 455. This report provides an exhaustive analysis of these technologies, examining their architectural mechanisms, benchmarking their performance, and evaluating the critical methodological limitations currently debated within the scientific community.
Generative Models for Crystal Structure Prediction
Crystal Structure Prediction (CSP) is the algorithmic task of determining the stable, three-dimensional spatial arrangement of atoms based solely on a specified chemical composition. Unlike the generation of finite organic molecules or proteins, CSP encounters distinct topological challenges due to the infinite, periodic nature of crystalline geometries. An effective generative model must account for strict symmetries, including translation, rotation, and periodic boundary conditions defined by the material's space group and lattice vectors 2. Over the past several years, diffusion-based architectures and fine-tuned large language models have emerged to address these unique spatial requirements.
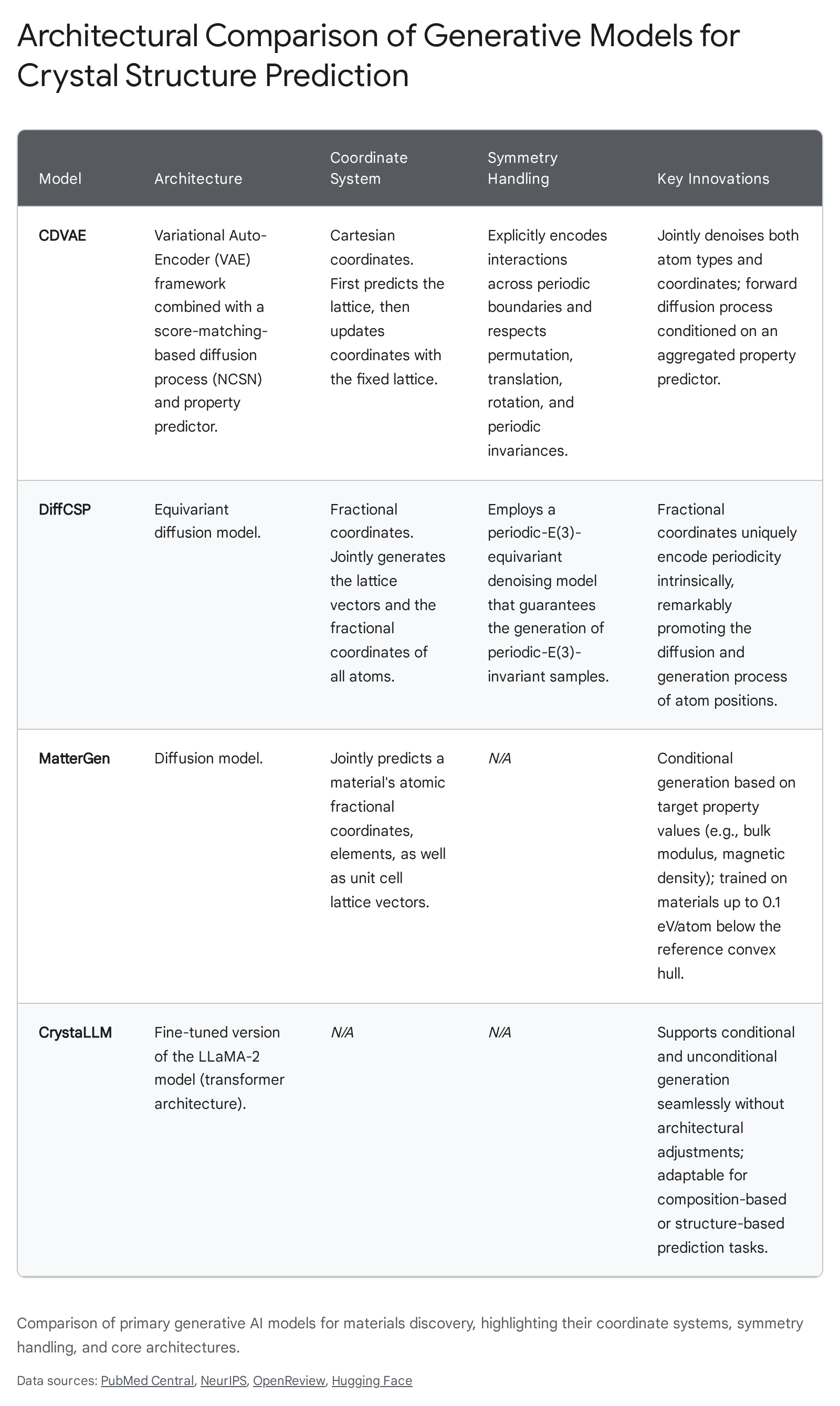
Diffusion Architectures and Coordinate Systems
Diffusion models generate data by progressively denoising a highly disordered state into a structured, physically viable output. Early applications of diffusion to CSP, such as the Crystal Diffusion Variational Autoencoder (CDVAE), utilized an equivariant variational autoencoder framework. CDVAE learns the data distribution of known stable materials and decodes them through a diffusion process that moves atomic coordinates toward a lower energy state while simultaneously updating atom types to satisfy local bonding preferences 6. CDVAE was trained on established datasets like Perov-5 (perovskites), Carbon-24 (carbon allotropes), and MP-20 (inorganic materials from the Materials Project), demonstrating a high capacity to generate realistic periodic structures 78. However, CDVAE traditionally handles structural generation sequentially - predicting the lattice first, then updating the atomic coordinates within that fixed lattice - and relies heavily on Cartesian coordinates, which can complicate the intrinsic representation of periodicity across boundaries 2.
To resolve the limitations of fixed lattices and Cartesian boundaries, researchers developed DiffCSP, an equivariant diffusion method that introduces joint generation. DiffCSP simultaneously diffuses both the lattice vectors and the atomic coordinates, treating the crystal geometry as a unified whole. A critical architectural choice in DiffCSP is the utilization of fractional coordinates rather than Cartesian coordinates. Because fractional coordinates describe atomic positions relative to the lattice vectors, they intrinsically encode periodic boundaries. DiffCSP employs a periodic-E(3)-equivariant denoising model, which guarantees that the intermediate distributions throughout the diffusion process remain invariant under rotation and translation 2. In direct benchmark comparisons on the MP-20 and Perov-5 datasets, models utilizing joint diffusion on fractional coordinates significantly outperform sequential methods in terms of Root Mean Square Error (RMSE) and match rates against ground-truth DFT structures 910.
Property-Conditioned Inverse Design
While the unconditional generation of stable crystals demonstrates that a model has successfully mapped the thermodynamic energy landscape, applied materials science requires "inverse design" - the ability to generate compounds that satisfy specific, predefined property constraints. Microsoft Research's MatterGen represents a highly advanced implementation of property-conditioned generation. Built on a diffusion framework that refines atom types, coordinates, and periodic lattices, MatterGen incorporates specialized adapter modules that allow the base model to be fine-tuned toward specific mechanical, electronic, or magnetic targets using labeled datasets 1112.
MatterGen was trained on over 600,000 stable materials derived from the Materials Project and Alexandria databases, ensuring a broad baseline understanding of chemical viability 13. The model's capacity for inverse design is evaluated using the S.U.N. metric - measuring whether generated materials are Stable, Unique, and Novel. When explicitly conditioned to generate structures with a bulk modulus greater than 400 GPa (indicating extreme physical hardness), MatterGen successfully proposed 106 S.U.N. structures within a budget of 180 DFT verifications. This performance is notable because only two such extreme-modulus structures existed in the reference training set 1114. Furthermore, MatterGen is capable of handling multi-property constraints simultaneously, such as targeting a high magnetic density (e.g., > 0.2 $A^{-3}$) while deliberately restricting the chemical composition to elements with low supply-chain risk profiles 1112.
Despite these capabilities, unconstrained diffusion processes frequently struggle to maintain strict crystallographic symmetry during the generative denoising phase. For instance, when researchers stipulate a target space group during conditioning, subsequent structural relaxation often results in a deviation from the desired crystal system 16. To mitigate this, newer symmetry-aware generative models, such as WyckoffDiff and Space-Group Conditional Flow Matching (SGFM), have been introduced. WyckoffDiff explicitly integrates symmetry priors - such as Wyckoff positions - directly into the diffusion input parameters, effectively constraining the generative process and preserving high-symmetry space groups through final structural optimization 916.
Large Language Models in Crystallography
Concurrent with the development of diffusion models, researchers are adapting Large Language Model (LLM) architectures for crystal generation. Models like CrystaLLM apply a customized version of the LLaMA-2 architecture to structural data representations 9. The primary advantage of LLM-based generative models is their inherent flexibility regarding conditioning. Unlike diffusion models, which typically require structural augmentation or specialized adapter modules to switch between conditional and unconditional tasks, LLMs handle both seamlessly through text-based prompting. CrystaLLM was evaluated on highly complex datasets, such as MPTS-52 (which contains up to 52 atoms per unit cell), demonstrating competitive RMSE scores against leading diffusion networks without requiring complex geometric priors 9.
Universal Machine Learning Interatomic Potentials (uMLIPs)
If generative models function as the architects proposing new atomic blueprints, machine learning interatomic potentials (MLIPs) are the physics engines required to simulate their operational behavior. Atomistic simulation has historically been divided between highly accurate but computationally restrictive Ab Initio Molecular Dynamics (AIMD) and computationally efficient but chemically rigid Physics-Based Potentials (PBP) 15. MLIPs bridge this divide by employing deep neural networks to learn the underlying quantum mechanical interactions from DFT datasets, subsequently predicting potential energies, atomic forces, and stresses with near-quantum accuracy at classical force-field speeds 1516. Recently, the focus has shifted from narrowly fitted models optimized for specific alloys to universal MLIPs (uMLIPs) trained across the entire periodic table.
Architectural Evolution: Invariant to Equivariant Networks
The predictive fidelity of a uMLIP is fundamentally determined by its mathematical representation of the local atomic environment during message passing. Early graph neural networks relied on invariant message passing, wherein the features transmitted between atomic nodes were scalar quantities that remained unchanged under 3D rotation and translation 17. Prominent invariant models include M3GNet, which incorporates three-body angular interactions within its graph architecture, and CHGNet (Crystal Hamiltonian Graph Neural Network) 1819. CHGNet advanced the invariant paradigm by incorporating atomic charge states as fundamental inputs. Pre-trained on the MPtrj dataset (comprising over 1.5 million structures spanning a decade of Materials Project calculations), CHGNet excels in charge-informed atomistic modeling, accurately predicting magnetic moments alongside standard force metrics 1920.
However, invariant architectures are fundamentally limited in their geometric expressivity. The field has since pivoted toward equivariant message passing neural networks (MPNNs). Equivariant models preserve directional data (vectors and higher-order tensors) during the message-passing phase, allowing the neural network to inherently capture the orientation and symmetries of atomic interactions 1721. The Message Passing Atomic Cluster Expansion (MACE) is a leading framework in this category. MACE integrates the symmetry-adapted basis of the Atomic Cluster Expansion (ACE) with equivariant graph neural networks 21. By generating high-body-order messages through tensor products, MACE captures complex many-body interactions locally without requiring exceptionally deep neural network layers 17. This architectural efficiency allows MACE to deliver fast, highly accurate simulations suitable for both bulk inorganics (MACE-MP-0) and complex organic molecular liquids (MACE-OFF23) 1522.
Scale and Scope of Foundation Models
Alongside architectural improvements, the scope of training data dictates the practical boundaries of uMLIP applicability. Two major foundation models - GNoME and MatterSim - illustrate the impact of extreme-scale data curation.
Google DeepMind's Graph Networks for Materials Exploration (GNoME) relies on an active learning flywheel. Beginning with structural baselines from the Materials Project, OQMD, and WBM databases, GNoME proposed new candidate structures, validated their stability via DFT, and fed the high-quality calculations back into its training corpus. This iterative process yielded an unprecedented dataset of 2.2 million new crystal structures, of which 380,000 are predicted to reside on or below the convex hull, indicating definitive thermodynamic stability 2324. The resulting dataset has enabled the training of robust interatomic potentials capable of zero-shot generalization, significantly accelerating the identification of highly specific functional materials, such as 528 potential new lithium-ion conductors 2324.
In contrast, Microsoft Research's MatterSim targets the profound limitations of models trained predominantly on 0 K ground-state relaxations. MatterSim is an atomistic emulator pretrained on 35 million first-principles-labeled structures explicitly sampled across vast thermodynamic conditions - ranging from 0 to 5000 Kelvin and ambient pressures up to 1000 GPa 252627. By mapping the potential energy surface far from equilibrium, MatterSim operates effectively for highly dynamic simulations involving melting, phase transitions, and amorphous solids. The latest iteration, MatterSim-MT (Multi-Task), extends predictions beyond energy and force to compute Bader charges, magnetic moments, and dielectric matrices natively 2528.
Dynamic Stability and Molecular Dynamics Benchmarks
While uMLIPs perform exceptionally well on static validation sets, their true utility is measured during finite-temperature molecular dynamics (MD) simulations. A comprehensive 2025 benchmark analyzed the performance of leading uMLIPs across the AMCSD-MD-2.4K dataset, which contains approximately 2,400 experimentally validated mineral structures 20. MD simulations expose a critical vulnerability in uMLIPs: when an algorithm explores an off-equilibrium region of configuration space where its force gradients are inaccurate, numerical errors accumulate rapidly, leading to explosive, non-physical structural divergence 1520.
The benchmark revealed stark disparities in the dynamic stability of different architectures. Equivariant models and non-equilibrium-trained models demonstrated remarkable resilience, whereas older invariant models struggled to complete standard simulation trajectories.
| Interatomic Potential Model | Architecture Type | MD Completion Rate | Density MAPE (%) | Lattice MAPE (%) |
|---|---|---|---|---|
| ORB | Equivariant MPNN | 99.96% | 8.67% | 3.33% |
| SEVENNET | Equivariant MPNN | 98.75% | 13.87% | $6.85 \times 10^{30}$%* |
| MatterSim | Graphormer / M3GNet | 85.00% (Approx) | 8.84% | 9.57% |
| MACE | Equivariant (ACE) | 80.00% (Approx) | 12.85% | $1.84 \times 10^{17}$%* |
| M3GNet | Invariant Graph | 15.00% | 76.29% | 191.00% |
| CHGNet | Invariant Graph | 7.00% | 46.97% | 77.20% |
Note: Extreme lattice deviations in SEVENNET and MACE correspond to localized simulation explosions within a subset of complex phase-space trajectories, though their overall completion and density retention remain high relative to invariant baselines 20.
Beyond dynamic stability, models are evaluated on their ability to predict derivative mechanical properties. In comparisons of bulk modulus predictions against theoretical data for 11,000 elastically stable materials, SEVENNET and MACE exhibited the highest consistency with DFT (Pearson correlation R $\approx$ 0.94, MAE $\approx$ 15 GPa), marginally outperforming MatterSim and significantly outperforming CHGNet 31. For highly sensitive variables such as the shear modulus, MACE and SEVENNET attained the highest correlations, whereas CHGNet systematically underestimated structural rigidity 31.
Autonomous Laboratories and Closed-Loop Synthesis
The exponential increase in computationally predicted materials creates a severe experimental bottleneck. Synthesizing, characterizing, and validating hundreds of thousands of candidate materials surpasses the physical bandwidth of traditional human-operated laboratories. To bridge this divide, research institutions are developing autonomous "self-driving laboratories" (SDLs). These platforms integrate AI-driven experimental design, robotic synthesis hardware, and high-throughput automated characterization to execute the predict-make-measure discovery loop without human intervention 529.
Specialized Robotic Platforms
USTC AI-Chemist: The University of Science and Technology of China (USTC) developed a highly sophisticated autonomous platform, represented by robotic agents named "Xiao Lai" and "Luke." The architecture relies on a multi-agent system comprising a Literature Reader, Experiment Designer, Computation Performer, and Robot Operator 29. The system processes vast quantities of existing literature (e.g., parsing 50,000 academic papers in two weeks) to establish protocol libraries 30. In a prominent demonstration, the AI-Chemist was tasked with discovering an oxygen-evolution reaction (OER) catalyst utilizing Martian meteorites. By analyzing the elemental composition of ores in real-time via laser-induced breakdown spectroscopy (LIBS), the AI performed density functional theory calculations to estimate catalytic activity across tens of thousands of high-entropy hydroxide ratios. The robotic system physically synthesized and evaluated the top candidates, identifying a stable catalyst capable of decomposing water at Martian temperatures with an overpotential of 445.1 mV. Researchers estimate this specific optimization would have required 2,000 years of manual human experimentation 3132. Furthermore, the USTC AI-Chemist was utilized to build a general mathematical theory of metal-support interactions (MSI) in heterogeneous catalysis, deriving a novel predictive formula from automated historical data analysis 33.
Berkeley Lab's A-Lab and AutoBot: The Lawrence Berkeley National Laboratory (LBNL) has aggressively pursued solid-state and thin-film automation. The A-Lab was designed as an autonomous solid-state powder synthesis facility. Utilizing generative recipes derived from the Materials Project and GNoME databases, the A-Lab employs robotic arms to weigh, mix, and heat precursor powders 534. In its inaugural 17-day autonomous run, the A-Lab claimed a 71% success rate, synthesizing 41 target inorganic compounds out of 58 attempts 3536. Complementing the solid-state A-Lab, LBNL developed AutoBot, a platform optimized for liquid-based thin-film synthesis. AutoBot utilizes high-speed active learning to navigate multi-dimensional synthesis parameters. When tasked with synthesizing high-quality metal halide perovskites (materials critical for advanced solar cells and LEDs), AutoBot pinpointed optimal relative humidity bounds (5% to 25%) by sampling merely 1% of the 5,000 possible parameter combinations, completing in weeks what would conventionally take a year 37.
VinUniversity AIRES Project: The synthesis of highly porous materials, such as Metal-Organic Frameworks (MOFs) and Zeolitic Imidazolate Frameworks (ZIFs), is notoriously difficult, with traditional intuitive success rates as low as 25%. VinUniversity, in collaboration with UC Berkeley, launched the Algorithmic Iterative Reticular Synthesis (AIRES) project to automate this process. AIRES utilizes Random Forest and Gaussian Process classifiers to navigate the probabilistic outcomes of complex chemical systems. Crucially, the platform integrates computer vision models capable of examining microscope imagery to identify successful crystallization with an 88% recall rate 3839. During an autonomous 700-experiment campaign, AIRES discovered 11 novel ZIF structures from 48 organic linkers, effectively doubling the efficiency of traditional methods 38.
Performance Metrics for Automation
As SDLs proliferate, researchers emphasize that traditional metrics, such as gross optimization rate, fail to capture the nuanced efficiency of automated platforms across different chemical domains 40. Complex experimental spaces inherently possess varying dimensionalities and noise floors. Consequently, contemporary evaluation frameworks are adopting statistical models like bagging regression and upper confidence bound decision policies to quantify Bayesian optimization efficiency under noisy conditions 40. Furthermore, laboratories are implementing Sigma Metrics to calculate defect rates per million opportunities across the total testing process, allowing operators to systematically evaluate hardware-software integration barriers and robotic dispensing accuracy 4142.
Methodological Limitations and Community Critique
Despite the unprecedented volume of theoretically stable materials proposed by models like GNoME and MatterGen, widespread adoption within the experimental chemistry community remains tempered by profound methodological skepticism. Rigorous peer review of highly publicized AI milestones has revealed structural vulnerabilities in how computational algorithms interpret phase stability and physical reality.
The Compositional Disorder Problem
The most substantial critique of recent AI discoveries targets the baseline assumptions of Density Functional Theory training data. Materials chemists Robert Palgrave and Leslie Schoop published a comprehensive analysis refuting the novelty of the 41 compounds synthesized by LBNL's A-Lab, concurrently questioning the utility of the millions of compounds within the GNoME database 3643.
The primary vector of failure involves compositional disorder. Generative models and their underlying DFT evaluators operate at an idealized 0 Kelvin. Because the models do not account for thermal entropy, the algorithms computationally force distinct elements into rigid, highly ordered crystallographic sites to achieve absolute minimum energy states 4344. However, physical solid-state synthesis requires extremely high temperatures. Under these thermal conditions, entropic effects dominate, causing different constituent elements to randomly share the same crystallographic sites. This physical reality results in highly symmetric, compositionally disordered solid solutions or common alloys 4344.
When Palgrave and Schoop analyzed the theoretically "novel" structures proposed by AI, they discovered that roughly two-thirds were simply mathematically ordered, 0 K approximations of well-known, disordered alloys that have existed in chemical literature for decades 4345. Critics argue that populating databases with hundreds of thousands of these artificially ordered phases dilutes the search space, creating an illusion of discovery while offering little functional utility to experimentalists seeking genuinely novel physical properties 444549.
Autonomous Characterization Failures
The critique extends beyond theoretical generation into the physical validation mechanisms of autonomous labs. In self-driving setups like the A-Lab, success is verified via automated Rietveld analysis of powder X-ray diffraction (XRD) data. Human crystallographic review of the A-Lab's autonomous output revealed that the AI's interpretation of XRD patterns was fundamentally unreliable 3643. The automated software frequently confused multi-phase mixtures and known alloys with the successful synthesis of the target compound, resulting in systemic false positives. The community stresses that until AI-driven XRD interpretation algorithms achieve the nuanced expertise of human crystallographers, autonomous "discoveries" remain highly suspect 43.
The Synthesizability Bottleneck
Generative AI models excel at mapping stability but frequently fail to account for kinetic synthesizability - whether a chemical pathway actually exists to construct the proposed crystal in a laboratory setting. Because the combinatorial space spans trillions of theoretical configurations, defining synthetic accessibility mathematically is highly complex. To address this, researchers are turning to Large Language Models pre-trained on materials data. For example, the LLM-Prop framework was fine-tuned using a novel sequence representation (MOFSeq) to predict the free energy and synthetic accessibility of Metal-Organic Frameworks. The model achieved a Mean Absolute Error (MAE) of 0.789 kJ/mol on free energy predictions and correctly predicted MOF synthesizability with a 97% F1 score without requiring retraining 50. Integrating synthesizability constraints directly into generative algorithms is considered the next critical milestone in model development.
Downstream Applications and Ecological Integration
While debates regarding absolute novelty persist, targeted applications of AI in materials science are undeniably accelerating the optimization of functional technologies, particularly in sustainable energy.
Energy Storage and Electrocatalysis
The transition to clean energy relies on identifying materials with specific electrochemical or thermodynamic properties. AI screening routinely narrows vast chemical spaces into viable experimental cohorts. For instance, researchers parsed the 380,000 stable crystals in the GNoME database to create Energy-GNoME, a curated subset identifying 33,000 compounds with high potential for energy applications. Using regressive machine learning tools to predict properties like the thermoelectric figure of merit ($zT$) and cathode voltage ($\Delta V_c$), the protocol highlighted over 21,000 potential battery cathodes and 4,200 novel perovskite configurations optimized for solar energy conversion 4652.
In advanced battery design, AI is facilitating a departure from conventional lithium-ion architectures. Utilizing DFT and AI modeling, researchers at USTC designed an anode-free Lithium-Hydrogen (Li-H) battery. By computationally mapping ion transport within the electrolyte, they developed a system that deposits lithium from salts directly during charging, achieving an exceptional round-trip efficiency of 99.7% and a theoretical energy density of 2825 Wh/kg 47.
Life Cycle Assessment (LCA) and Safe-by-Design
As generative models propose materials lacking industrial precedent, ensuring their environmental safety is critical. The integration of environmental Life Cycle Assessment (LCA) with machine learning represents a major advancement in "safe-by-design" methodologies. The ML-LCA framework couples machine-learned interatomic potentials with global environmental databases to predict the ecological impact, human toxicity, and energy requirements of a material directly from its atomic characteristics 4849. By utilizing surrogate models to estimate scale-up manufacturing constraints, algorithms can co-optimize a material's functional performance (e.g., energy density or catalytic efficiency) alongside its environmental footprint, preventing the commercialization of highly efficient but ecologically toxic compounds 49.
Conclusion
Artificial intelligence is comprehensively transforming the operational scale and speed of materials science. Generative frameworks, evolving from Cartesian autoencoders to equivariant, fractional-coordinate diffusion models, are successfully mapping the thermodynamic boundaries of inorganic chemistry. Correspondingly, universal machine learning interatomic potentials, trained on tens of millions of structural evaluations, provide the essential physics engines to simulate these materials at finite temperatures with near-quantum accuracy. The deployment of self-driving laboratories signifies the critical next step, successfully automating the translation of computational predictions into physical compounds.
However, the field remains in a state of recalibration. Rigorous critiques of massive theoretical databases highlight that algorithmic generation without strict physical constraints - particularly regarding high-temperature entropy and compositional disorder - often yields artifacts rather than applicable discoveries. The immediate future of AI in materials science will rely less on expanding raw candidate volume and more on refining the interface between targeted, property-conditioned generative modeling and robust, human-validated robotic synthesis. By integrating synthesizability metrics and lifecycle sustainability directly into the predictive loop, AI is poised to transition from theoretical exploration to the reliable industrial deployment of advanced functional materials.