Antagonistic pleiotropy theory of aging
Foundations of Evolutionary Aging Theories
The persistence of aging and senescence across nearly all multicellular life presents a fundamental paradox in evolutionary biology. If natural selection rigorously favors traits that enhance survival and reproduction, the near-universal age-related deterioration of physiological function and exponentially increased mortality risk appears mathematically counterintuitive. To resolve this paradox, mid-twentieth-century evolutionary biologists established the foundational theories of the evolution of aging, shifting the focus from aging as a genetically programmed, species-level inevitability to aging as a byproduct of the declining force of natural selection over an individual organism's lifespan 12.
The framework for this understanding was initiated by Peter Medawar in 1952 with the mutation accumulation theory. Medawar posited that because wild populations experience high rates of extrinsic mortality driven by predation, disease, and starvation, statistically few individuals survive to advanced ages in natural ecosystems. Consequently, the force of natural selection is profoundly weakened in late life, creating a theoretical "selection shadow" 12345. Mutations that express deleterious effects only at an advanced age are effectively invisible to natural selection and gradually accumulate in the population via genetic drift, ultimately manifesting as the physiological decline recognized as senescence 1356.
Building directly upon Medawar's concept of the selection shadow, George C. Williams formalized the antagonistic pleiotropy hypothesis in 1957 175610. Pleiotropy refers to the established genetic phenomenon in which a single gene or genetic locus influences multiple, seemingly unrelated phenotypic traits 15. Williams hypothesized that certain pleiotropic genes confer a strict evolutionary advantage by heavily enhancing fitness - defined primarily by survival to maturity and early reproductive success - early in the life course. However, these identical genes exhibit antagonistic, deleterious effects late in life 1256. Because the strength of natural selection is exponentially higher during the prereproductive and early reproductive phases compared to the post-reproductive phase, alleles with robust early-life benefits will be strongly favored by directional selection and fixed in the population, even if they directly cause catastrophic health deterioration in old age 125.
Under the antagonistic pleiotropy framework, aging is not an adaptive trait selected for its own biological utility, but rather an inescapable biological trade-off. Natural selection acts strictly to maximize lifetime reproductive success, not indefinite somatic maintenance or longevity 78. Therefore, the physiological and cellular mechanisms driving senescence are inextricably linked to the precise mechanisms that drive early-life growth, metabolic development, and optimal fertility 789.
Mathematical Formalization of Natural Selection
To rigorously evaluate the antagonistic pleiotropy hypothesis, evolutionary biologists rely on quantitative models of life-history traits and selection pressure. William D. Hamilton mathematically formalized the declining force of natural selection in 1966 through indicators that measure the sensitivity of overall population growth to specific, age-dependent changes in survival and fertility parameters 3101116.
Hamiltons Indicators and Population Dynamics
Hamilton utilized the intrinsic rate of population increase ($r$), derived from the discrete Lotka equation, which integrates the age-specific survival function ($l_x$) and the age-specific maternity or fertility function ($m_x$) 1011. Hamilton demonstrated that the selective evolutionary value of a genetic mutation depends almost entirely on the specific age at which its phenotypic effects are expressed. For a mutation altering survival at age $x$, the force of selection is strictly proportional to the remaining expected reproductive output of the individual from that age onward, often referred to as Fisher's reproductive value 6101112.
Because the survival probability $l_x$ invariably declines over chronological time due to baseline environmental hazards, and the reproductive function $m_x$ eventually reaches zero in senescing organisms (or naturally plateaus), the mathematical sensitivity of organismal fitness to age-specific mortality must strictly decline after the onset of sexual reproduction 1011. For instance, in theoretical models simulating high-extrinsic mortality populations, cohort survival can easily fall to 0.25% by age 34, rendering selection mathematically impotent beyond that threshold 1011.
Alternative Parameterizations and Selection Gradients
Subsequent researchers have expanded on Hamilton's indicators, demonstrating that while the core premise holds, the exact shape of the selection gradient depends heavily on the parameterization of mutational effects (e.g., additive versus proportional survival changes) and species-specific reproductive timelines 1314. Some alternative indicators suggest that the force of selection can theoretically increase leading up to peak reproductive age before commencing its terminal decline 161314.
Furthermore, the integration of parental care and intergenerational resource transfers alters the basic mathematical assumptions. In highly social species, such as humans, the force of selection does not necessarily drop to absolute zero immediately following reproductive cessation 1612. The "Grandmother Hypothesis" suggests that post-reproductive longevity is maintained because elder individuals contribute substantial caloric surpluses and care to descendants, thereby increasing their inclusive genetic fitness long after their own fertility has ended 1215. Empirical data indicates that while chimpanzees retain only 21% of their lifetime productive capacity after age 30, human hunter-gatherer populations retain up to 66% of their productive value at the same age, extending the reach of purifying selection and modifying the shape of the selection shadow 512.
Demographic Transitions and Selection Pressure
The mathematical boundaries of antagonistic pleiotropy rely heavily on the demographic environment. Recent demographic transitions - characterized by historically unprecedented reductions in both extrinsic mortality and fertility over the past century - have dramatically reshaped the human selective environment on a global scale.
Modernity and the Steepening Selection Gradient
An extensive demographic analysis spanning 175 countries over a 74-year period (1950 to 2023) mapped changes in Hamilton's force-of-selection framework 5. The data revealed an "extension-dilution trade-off" in modern populations. While the age at which selection intensity drops to half its maximum extended by 1.7 years, the overall peak selection intensity at birth declined by 29.4% due to drastically reduced childhood mortality 5.
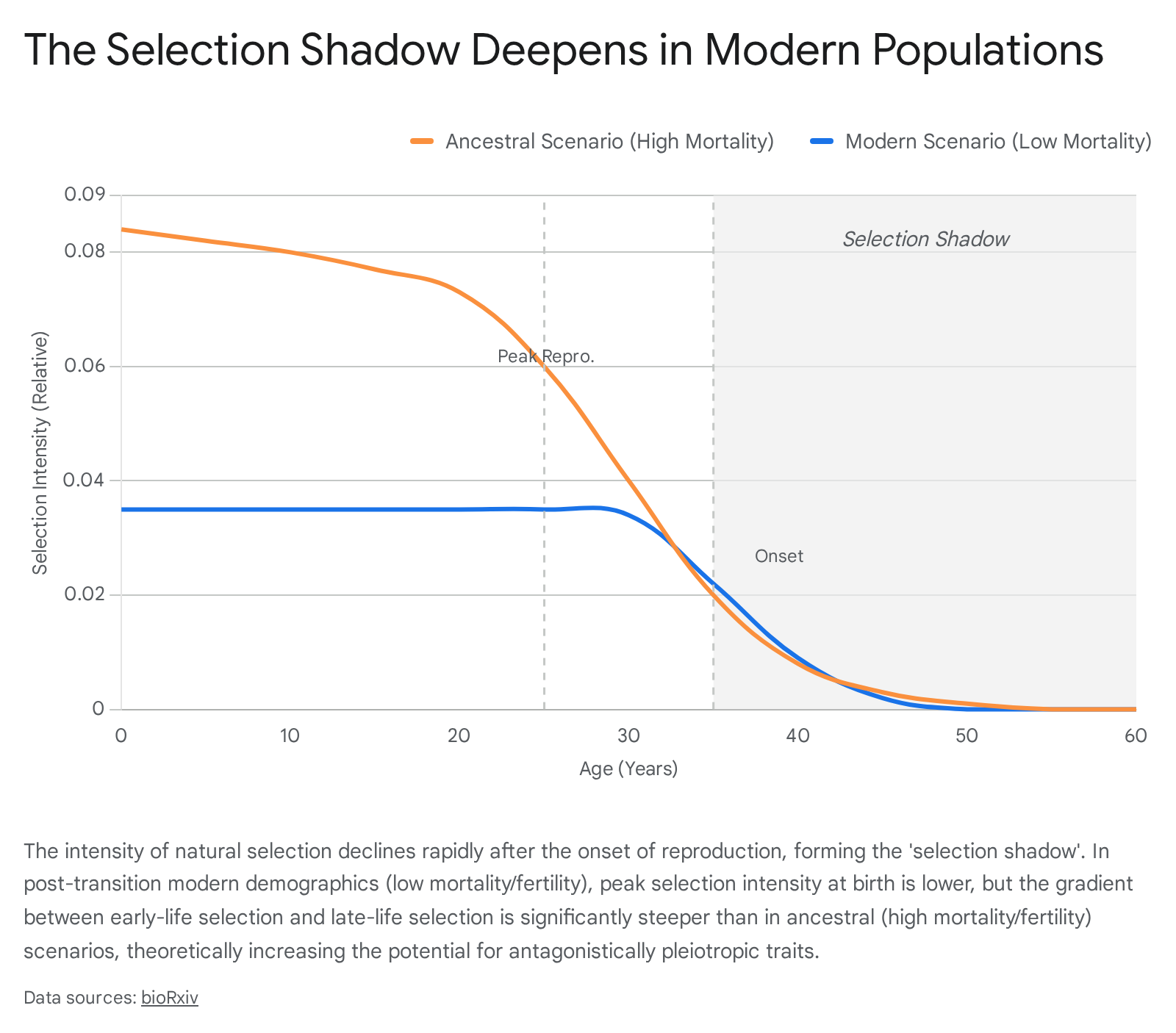
Crucially, antagonistic pleiotropy is driven not by absolute selection strength, but by the relative gradient between early and late life. The analysis demonstrated that this gradient has steepened sharply. The ratio of selection intensity at age 20 relative to intensity at age 40 rose from 17.3 in ancestral regimes to 25.1 in post-transition demographics 5. This steeper gradient significantly amplifies the theoretical potential for antagonistic pleiotropy. Alleles conferring early-life reproductive or developmental benefits now face proportionally much stronger selection relative to their late-life pathological costs 5.
Summary of Selection Gradient Shifts
| Demographic Regime | Mortality/Fertility Profile | Selection Intensity at Birth $s(0)$ | Selection at Age 40 $s(40)$ | Selection Ratio $s(20)/s(40)$ | Antagonistic Pleiotropy Potential |
|---|---|---|---|---|---|
| Ancestral (Pre-Transition) | High Mortality / High Fertility | 0.084 | 0.008 | 17.3 | Baseline / Moderate |
| Transitional | Moderate Mortality / Early Fertility | 0.071 | 0.003 | 21.3 | High |
| Modern (Post-Transition) | Low Mortality / Low Fertility | 0.035 | 0.009 | 25.1 | Exceptionally High |
Data reflects historical averages and recent trends demonstrating the mathematical steepening of the selection gradient 5.
Because modern environmental stability allows humans to routinely function for decades beyond ancestral baselines, the severe late-life costs of antagonistically pleiotropic alleles are now manifesting at unprecedented scales, while the evolutionary pressure to organically maintain late-life somatic integrity has never been weaker 5.
Genomic Evidence in Human Populations
For decades following Williams's initial publication, antagonistic pleiotropy remained a mathematically sound but difficult-to-prove theoretical construct. Identifying specific genetic loci that definitively modulate both early-life reproductive fitness and late-life mortality requires massive sample sizes, deeply phenotyped individuals, and robust longitudinal mortality tracking 516. The recent advent of population-scale genomic repositories has provided unambiguous, genome-wide evidence supporting the widespread existence of antagonistic pleiotropy in humans 617.
Tradeoffs Between Reproductive Traits and Lifespan
A definitive genomic test of the hypothesis was conducted by Long and Zhang in 2023, utilizing whole-genome data, reproductive phenotypes, and death registry records from 276,406 participants in the UK Biobank 61623. The researchers specifically investigated the genetic correlation between highly heritable reproductive traits - such as age at first birth, age at first sexual intercourse, and total number of children fathered or mothered - and parental lifespan, utilizing survival to age 76 (SV76) as a benchmark 6.
The analysis revealed a powerful, negative genetic correlation between reproductive output and overall lifespan across both sexes 616. Individuals carrying higher polygenic scores for reproduction (PGSR) exhibited systematically lower probabilities of survival. For instance, participants ranked in the top third for a polygenic score driving an earlier age at first birth had a survival probability to age 76 of 0.800, compared to a significantly higher survival probability of 0.839 for those in the bottom third of the score 623.
Variant-level data yielded starker evidence. Of 583 total genetic polymorphisms found to be strongly associated with reproductive traits, 123 significantly impacted overall lifespan 6. Among these, 98 variants exhibited pure antagonistic pleiotropy (the exact allele enhancing reproduction actively reduced lifespan), compared to only 25 variants that showed concordant pleiotropy (where the allele benefited both) 623. Statistical modeling confirmed that genetic polymorphisms impacting reproduction were 7.5 times more likely to exert an antagonistic, lifespan-shortening effect than random baseline polymorphisms 62318.
Importantly, the researchers established that this correlation stems from "horizontal pleiotropy" rather than the mere physical toll of childbearing. When statistically controlling for the total number of children born to an individual, the negative correlation between reproductive polygenic scores and survivorship remained robust 618. This indicates that the genetic variants dictate both reproduction and lifespan through separate, intrinsic biochemical pathways (e.g., systemic hormonal signaling or cellular proliferation rates) rather than through the direct mechanical and physiological exhaustion of the reproductive act itself 6. Molecular analysis of these antagonistically pleiotropic variants indicated that they predominantly influence cis-regulatory activity across multiple target genes and tissues, heavily enriched in pathways managing estrogen receptor signaling, immune response, and amyloid processing 6.
Additionally, the researchers detected active, ongoing natural selection for these traits. The frequencies of reproduction-enhancing, lifespan-shortening alleles measurably increased across successive UK Biobank birth cohorts spanning from 1940 to 1969. This data confirms the evolutionary prediction that natural selection continues to actively favor early-life reproductive output at the direct expense of post-reproductive health 618.
Pleiotropy in Cardiometabolic and Complex Diseases
The imprint of antagonistic pleiotropy is highly visible in the genetic architecture of specific, age-related human pathologies. A foundational 2017 study by Byars et al. investigated the evolutionary genetics underpinning coronary artery disease (CAD), a leading global cause of late-life mortality 1920. The research tested whether loci associated with CAD showed evidence of evolutionary selection based on early-life reproductive traits.
The findings revealed that genetic loci driving CAD are significantly enriched for traits linked to lifetime reproductive success (LRS). Out of 76 established CAD-associated genes, 51 contained single nucleotide polymorphisms that were positively associated with increased LRS 20. Crucially, the relationships proved deeply antagonistic in clinical cohorts like the Framingham Heart Study. Specific alleles in genes such as FLT1 (rs9319428-A) and LPA (rs2048327-C) significantly increased both overall reproductive success and the lifelong risk for CAD 20.
Further systematic reviews of the top 40 CAD genes showed that 100% of these loci shared pleiotropic connections to vital early-life fitness traits, including overall reproductive capacity, increased twinning probabilities, optimized age at menarche or menopause, and enhanced lactation capabilities 20. Because the pathological symptoms of CAD generally manifest well into the post-reproductive decades, the intense selective pressure for optimal early-life reproductive performance utterly overwhelms the weak, late-life purifying selection against cardiovascular degeneration 62021.
Beyond cardiovascular networks, broad investigations utilizing massive clinical datasets continue to map the antagonistically pleiotropic nature of aging. Studies leveraging Mendelian randomization on UK Biobank data confirm that a later age of menarche or first childbirth is genetically associated with a longer parental lifespan, delayed systemic epigenetic aging (as measured by DNA methylation clocks like GrimAge), reduced frailty indices, and lower incidence risks for late-onset Alzheimer's disease, heart failure, and specific chronic kidney diseases 2223.
Ancestral Defenses and Modern Pathologies
Patterns of extreme pleiotropy are distributed across diverse ethnic populations, often reflecting intense, localized evolutionary pressures. A comprehensive 2024 genetic analysis leveraging the VA Million Veteran Program examined the architecture of 2,068 traits across 347,000 individuals of diverse ancestries 17. The study mapped 15,596 trait-gene associations, confirming widespread horizontal pleiotropy.
The study identified the APOE gene as the most highly pleiotropic locus in the human genome, securely linked to 29 independent traits including Alzheimer's dementia, macular degeneration, and previously unrecognized associations with chronic liver disease 17. The APOE e4 variant is historically known to confer enhanced early-life cognitive development and robust resistance to endemic infectious agents, counterbalanced by a severe late-life neurodegenerative toll 91724.
The Million Veteran Program data also highlighted severe population-specific pleiotropic outliers driven by regional pathogen loads. In populations of African descent, genes such as APOL1, HBB, and CD36 exhibit massive pleiotropy compared to European cohorts 17. These loci were positively selected to confer critical early-life survival advantages against lethal parasitic infections, specifically malaria and trypanosomiasis. However, these identical survival alleles predispose carriers to severe late-life pathologies, including aggressive chronic kidney disease, sickle cell complications, and gout, perfectly illustrating the environment-dependent trade-offs central to the theory 117.
Similar evolutionary trade-offs are implicated in rising modern epidemics. For instance, genomic studies estimate that natural selection actively adds over 100,000 myopia cases per generation in the UK alone. The rapid spread of myopia risk alleles is primarily attributed to their statistically pleiotropic association with enhanced reproductive output, allowing a seemingly disadvantageous vision trait to hitchhike to high population frequencies on the back of early-life fitness advantages 31.
Summary of Key Antagonistically Pleiotropic Genes
| Genetic Locus | Primary Early-Life Selective Benefit | Late-Life Pathological Cost / Disease | Source Focus |
|---|---|---|---|
| FLT1 / LPA | Enhanced lifetime reproductive success | Severe Coronary Artery Disease (CAD) | 20 |
| APOE (e4) | Improved early cognitive performance, high fertility | Alzheimer's dementia, Cardiovascular decline | 91724 |
| HBB | Resistance to malarial infections | Adult-onset gout, Sickle cell pathology | 117 |
| Slc2a4 (GLUT4) | Aggressive cellular glucose uptake for rapid growth | Neural stem cell exhaustion in the aging brain | 25 |
| KAT7 | Chromatin regulation for active cellular proliferation | Systemic cellular senescence, Progeroid syndromes | 33 |
Cellular and Molecular Mechanisms
While Genome-Wide Association Studies (GWAS) provide statistical validation of antagonistic pleiotropy, understanding how these genetic trade-offs physically execute within tissues requires molecular mapping. The cellular manifestation of these evolutionary compromises is increasingly understood through highly conserved nutrient-sensing, metabolic, and epigenetic networks 22627.
Hyperfunction Theory and Anabolic Pathways
The Hyperfunction Theory of aging, advanced prominently in contemporary geroscience, provides a precise molecular mechanism for antagonistic pleiotropy 242829. Classical models assumed aging was driven by "hypofunction" - the passive, mechanical wear-and-tear and accumulated biochemical damage over time 330. In sharp contrast, the Hyperfunction Theory posits that aging is actively driven by the relentless, programmatic overactivity of critical developmental pathways that lack an evolutionary "off switch" 24282930.
Central to this model is the highly conserved anabolic metabolic axis, including the mechanistic target of rapamycin (mTOR), Insulin-like Growth Factor 1 (IGF-1), Growth Hormone (GH), and downstream kinases 2428. These cellular signaling networks are absolutely essential for orchestrating rapid embryonic development, childhood growth, and the attainment of sexual maturity. Consequently, they are under immense positive selection early in life 2428.
However, once developmental and somatic maturity is reached, these pathways do not organically deactivate. Due to the selection shadow, there is negligible evolutionary pressure to evolve complex regulatory mechanisms to downregulate these genes in later life 242830. Thus, the sustained, inappropriate signaling of mTOR and IGF-1 in adulthood drives cellular hypertrophy, hyperplasia, stem cell exhaustion, and the accumulation of senescent cells, culminating in systemic pathologies like cancer, organ hypertrophy, and metabolic exhaustion 242830. Under this paradigm, diseases of aging are not the result of failed biological maintenance, but the direct consequence of unchecked developmental momentum operating out of context 24282930.
Epigenetic Drift and Information Loss Models
Beyond hard-coded metabolic pathways, antagonistic pleiotropy is actively mapped in the epigenome. Epigenetic drift - the gradual, progressive loss of precise DNA methylation and chromatin organization over time - is a fundamental hallmark of aging across mammalian species 2731.
Information-theoretic models of aging suggest that differentiated cells actively slow epigenomic degradation through sustained, highly costly resource investment in repair mechanisms 27. In early life, robust epigenetic "cleanup" mechanisms operate efficiently, yielding massive survival benefits. However, because these repair genes are resource-intensive, they are inherently constrained by the energetic trade-offs required for reproduction 27. As epigenetic drift progressively degrades the very pathways responsible for genomic maintenance, accumulating transcriptional waste overwhelms the system, generating pathology 27. The failure occurs exclusively in late life, while the survival benefits persist through the reproductive peak, ensuring the energetic limitation strategies remain fixed in the population by natural selection 27.
Biomarkers of Aging and the Smurf Phenotype
The distinct shift from functional early-life states to late-life pathological states is visually and biologically quantifiable in model organisms. In Drosophila melanogaster, a well-documented biomarker of this transition is the "Smurf phenotype," characterized by a sudden, catastrophic increase in intestinal permeability 4. The onset of this phenotype acts as a highly accurate predictor of natural death, demarcating a clear physiological phase transition.
The discovery of the Smurf phenotype in Drosophila, alongside comparable phenomena in nematodes, zebrafish, and mice, challenges models of aging as a strictly continuous, linear decline 4. Instead, it supports a framework where aging is marked by distinct phases driven by the systemic collapse of barrier functions. The evolutionary tolerance for this late-life barrier collapse directly aligns with antagonistic pleiotropy, as the genes governing early-life intestinal rapid cellular turnover and nutrient absorption eventually drive terminal tissue failure in the post-reproductive phase 4.
Functional Dissection via CRISPR Screening
Historically, moving from statistical associations to definitive molecular proof of antagonistic pleiotropy was hindered by the inability to precisely isolate and manipulate individual gene functions in aging cells. The development of high-throughput CRISPR-Cas9 genome editing and CRISPR interference (CRISPRi) platforms has fundamentally overcome this barrier, allowing researchers to systematically knock out or repress specific genes and observe the resultant physiological trade-offs in real-time 25323334.
Rejuvenation of Neural Stem Cells
A groundbreaking 2024 investigation utilized in vitro and in vivo genome-wide CRISPR-Cas9 screens to dissect the genetic drivers of regenerative decline in the mammalian brain 25. Aging dramatically impairs the ability of neural stem cells (NSCs) to transition from a state of quiescence to active proliferation, resulting in a systemic decrease in neurogenesis and highly defective repair mechanisms following injury 25.
By screening primary cultures of both young and old murine NSCs, researchers identified over 300 specific gene knockouts that possessed the unique ability to specifically restore activation capacity in old NSCs, without altering the functionality of young cells 725. Among the most potent interventions was the targeted knockout of the Slc2a4 gene, which encodes the GLUT4 glucose transporter 725.
The identification of Slc2a4 perfectly illustrates antagonistic pleiotropy at the cellular level. In early life and development, aggressive cellular glucose uptake is absolutely vital for rapid neurogenesis, synaptogenesis, and brain growth. However, the CRISPR screen revealed that as the organism ages, glucose uptake inappropriately increases in NSCs, overwhelming their metabolic capacity and forcing them into a permanent state of quiescence 725. By utilizing CRISPR to knock out Slc2a4, or by inducing transient glucose starvation, researchers effectively rejuvenated the aged NSCs, allowing them to proliferate and produce new neurons 725. An essential nutrient-sensing mechanism, optimized by natural selection for early developmental growth, becomes fundamentally maladaptive and hyperfunctional in late life, directly driving tissue senescence 725.
Modulating Replicative Senescence and Inflammaging
Parallel high-throughput studies utilizing CRISPR interference (CRISPRi) have isolated distinct genetic drivers responsible for different modalities of aging, specifically separating replicative senescence from "inflammaging" (the chronic, sterile, low-grade inflammation that pervades aged tissues) 33.
By performing genome-wide transcriptional repression screens on human primary adipose-derived mesenchymal stem cells (MSCs), researchers identified separate genetic networks governing these pathways 33. Utilizing a positive selection methodology, the screens identified genetic perturbations that allowed cells to persist longer and resist entering permanent cell-cycle arrest under chronic, growth-limiting conditions 33.
The screens identified powerful novel regulators of inflammaging, most notably CMKLR1 and SERPING1 33. Integrating the functional CRISPRi data with large-scale GWAS datasets (encompassing over 400 aging-related studies) confirmed that these specific inflammatory aging signatures map directly to diverse human aging processes across multiple organ systems 33. Targeted repression of these genes reversed the pro-inflammatory microenvironment driven by interleukin-6 (IL-6), significantly rejuvenating the cellular phenotype and providing precise novel targets for therapeutic modulation 33.
Epigenetic Resetting via Targeted Intervention
The application of CRISPR-Cas9 has also pinpointed epigenetic regulators functioning as antagonistically pleiotropic drivers of aging. Extensive screening arrays identified the KAT7 gene, an epigenetic modifier involved in chromatin regulation and cellular proliferation, as a primary promoter of cellular senescence in adult human stem cells 33.
While KAT7 function is necessary for baseline genomic regulation and cell cycling during organismal development, its continued activity in late-life stages actively promotes systemic senescence. Experimental inactivation of KAT7 via CRISPR methodologies yielded profound results. It successfully rejuvenated human cells exhibiting premature aging characteristics derived from patients with severe progeroid syndromes (Hutchinson-Gilford progeria and Werner syndrome) 33. Furthermore, when applied in vivo, the targeted immobilization of KAT7 significantly extended the lifespan and healthspan of both prematurely aging and naturally aging wild-type mice 33.
Evolutionary Constraints and Competing Models
While antagonistic pleiotropy provides a robust structural framework for the evolutionary genetics of aging, it operates concurrently with other established evolutionary paradigms. Fully interpreting aging requires integrating antagonistic pleiotropy with models of biological constraint and resource allocation 2335.
The Disposable Soma Theory
Formulated by Thomas Kirkwood in 1977, the Disposable Soma Theory explicitly addresses physiological and energetic trade-offs, often acting as the metabolic mechanism through which antagonistic pleiotropy is executed 9152330. The theory posits that all organisms operate within environments containing strictly finite metabolic and caloric resources. Natural selection forces a zero-sum trade-off between the energy an organism invests in somatic maintenance (e.g., highly accurate DNA repair mechanisms, robust antioxidant defenses, and perfect proteostasis) and the energy invested in somatic growth and direct reproduction 152330.
Because the "selection shadow" guarantees that an organism in the wild is highly unlikely to survive indefinitely due to extrinsic mortality (predation, harsh winters, famine), investing massive caloric energy into achieving perfect, immortal somatic maintenance is an evolutionary dead end 1536. The mathematically optimal evolutionary strategy is to invest heavily in early reproduction and rapid growth, allowing the "disposable soma" to be maintained only well enough to ensure reproductive success, after which it gradually accumulates molecular damage and degrades 152330. Where antagonistic pleiotropy focuses on specific genes with dual temporal functions, the disposable soma theory provides the overarching energetic architecture explaining why those genes were selected: investing in perfect longevity simply costs too much reproductive potential 152330.
Interconnection Constraints and Evolutionary Spandrels
Recent advancements in aging theory integrate antagonistic pleiotropy with concepts of strict biological constraint and "evolutionary spandrels" 3035. As biological systems evolved increasing complexity, functional networks became deeply intertwined. This high degree of interconnectedness creates profound constraints on the simultaneous optimization of coupled traits, meaning it is often biologically impossible to mutate a gene to remove a late-life cost without simultaneously destroying its essential early-life function 3035.
Aging pathologies can thus be viewed as "bad spandrels" - inevitable, unselected byproducts of the complex architectural constraints required to build a functional, reproducing organism 3035. A gene driving rapid calcium deposition is necessary for building a strong skeletal framework in youth; the architectural constraint dictates that the same biochemical pathway will inevitably drive pathological arterial calcification in old age 630. The biological impossibility of decoupling these functions ensures the trait remains fixed 3035.
Summary of Evolutionary Aging Theories
| Theory | Primary Proponent | Core Mechanism | Driver of Aging | Relationship to Antagonistic Pleiotropy (AP) |
|---|---|---|---|---|
| Mutation Accumulation | Peter Medawar (1952) | Genetic Drift | Accumulation of purely deleterious, late-acting mutations | Precursor to AP; operates strictly within the selection shadow 1235. |
| Antagonistic Pleiotropy | George Williams (1957) | Directional Selection | Genes selected for early-life fitness exert late-life pathological costs | The core genetic tradeoff model 1275. |
| Disposable Soma | Thomas Kirkwood (1977) | Energetic Allocation | Sub-optimal investment in somatic maintenance to maximize reproduction | The metabolic/resource framework through which AP is executed 152330. |
| Hyperfunction Theory | Mikhail Blagosklonny | Pathway Overactivity | Failure to deactivate essential developmental and anabolic pathways | A specific mechanistic subset of AP driving cellular overgrowth 2428. |
Clinical Translation and Future Therapeutics
The realization that human aging is fundamentally driven by antagonistically pleiotropic gene networks has initiated a profound paradigm shift in modern geroscience and clinical medicine. Historically, medical interventions have attempted to treat devastating age-related diseases - such as Alzheimer's, cardiovascular atherosclerosis, and severe osteoarthritis - as isolated, independent pathological entities 1937. The evolutionary medicine perspective dictates that these seemingly disparate diseases are simply varied, tissue-specific symptoms of a singular, underlying evolutionary trade-off 192137.
Because the physiological drivers of these diseases are genes that were absolutely essential for early-life embryonic development and sexual maturation, systemic, lifelong pharmacological inhibition of these biological pathways is inherently dangerous 19. However, the late-onset nature of these pathologies presents a unique therapeutic window. Because the early-life developmental benefits of antagonistically pleiotropic genes have already been safely realized in adult human patients, it is biologically viable to attenuate, repress, or modify these genetic pathways in later life without incurring the severe early-life fitness penalties that originally constrained their evolution 243438.
CRISPR Applications in Age-Related Pathologies
Advanced gene-editing technologies, notably CRISPR-Cas9, base editing, and prime editing (which allows for sequence replacement without creating genotoxic double-strand DNA breaks), are actively bypassing evolutionary constraints 32343839.
Clinical trials utilizing highly precise in vivo CRISPR therapeutics are currently underway. For example, biopharmaceutical trials are aggressively targeting the ANGPTL3 and PCSK9 genetic pathways 2040. Individuals carrying specific natural variants of ANGPTL3 exhibit significantly reduced lipid levels and a drastically lowered risk of coronary artery disease 40. By utilizing CRISPR to edit these specific loci in adult patients, researchers aim to permanently lower cardiovascular risk. Advanced delivery systems, such as highly specific ionizable lipid nanoparticles (LNPs) that exploit natural liver tropism through apolipoprotein E (ApoE) binding, are allowing researchers to safely target these CRISPR packages directly to hepatic tissues 34404142.
In advanced preclinical models, CRISPR therapies are being deployed to knock out specific oncogenes, mitigate cellular senescence, and correct age-related genomic instability 3234. Protocols are targeting the hyperfunctional accumulation of amyloid-beta by knocking out APP and PSEN1 genes in Alzheimer's models, while actively correcting inherited mutations in LRRK2 and PINK1 to restore mitochondrial function in Parkinson's disease models 34.
Environmental Mismatch and Demographic Implications
The application of targeted gene editing arrives at a critical juncture in human history. The biological consequences of antagonistic pleiotropy are highly sensitive to the surrounding environmental context, generating deep "evolutionary mismatches" 3743.
For the vast majority of hominid history, human genetic architecture evolved in ecosystems characterized by extreme pathogen loads, physical trauma, and chronic caloric scarcity 212337. Natural selection fiercely favored "thrifty" alleles that promoted aggressive fat storage, robust inflammatory responses, and rapid coagulation to survive famines and massive infections 23. The transition to modernity - marked by the eradication of major infectious diseases, sanitation, and the advent of highly processed diets and sedentary lifestyles - has generated a toxic mismatch 643. The hyper-active immune alleles selected for ancestral parasite clearance now drive modern epidemics of autoimmune disorders and the chronic systemic "inflammaging" underpinning neurodegeneration 33.
The targeted modulation of antagonistically pleiotropic genes via CRISPR and epigenetic reprogramming holds the potential to systematically correct these deep evolutionary mismatches 384453. By resetting cellular tissues to a more youthful functional state without inducing stem-cell pluripotency, longevity therapeutics aim to directly extend the human healthspan 3844. While profound ethical, regulatory, and demographic questions remain regarding access, inequality, and the boundary between disease therapy and human enhancement, reversing the pathological late-life consequences of antagonistic pleiotropy represents the most biologically grounded strategy for combating the systemic physiological decline of human aging 3353.