AI diffusion models for protein design
The transition of molecular biology from a predominantly observational science to a programmable engineering discipline has been fundamentally accelerated by the advent of artificial intelligence (AI). Historically, protein engineering was restricted to modifying naturally occurring amino acid sequences through directed evolution or rational structural design, thereby limiting researchers to exploring an exceptionally minuscule fraction of the theoretical sequence space 122. The theoretical design space of a moderately sized protein consisting of 100 amino acids exceeds the number of atoms in the observable universe, making brute-force computational searches impossible 34. Today, deep learning frameworks - particularly generative diffusion models, graph neural networks, and hybrid transformer architectures - enable the de novo design of entirely novel proteins that do not exist in nature 367.
These models generate custom amino acid sequences programmed to fold into specific three-dimensional topologies and execute targeted biochemical functions. Such functions range from binding to disease-associated receptors with picomolar affinity to neutralizing viral entry, scaffolding novel enzymes, or catalyzing industrial reactions 3475. This report provides an exhaustive analysis of generative AI in protein design, focusing on the mathematical foundations of diffusion models operating over three-dimensional manifolds. It evaluates the architectural differences between leading systems such as RFdiffusion, AlphaProteo, and Chroma, and outlines the end-to-end computational pipeline required to translate in silico designs into wet-lab validated therapeutics. Furthermore, the analysis addresses critical bottlenecks in modeling conformational flexibility, the profound biosecurity risks posed by AI-generated pathogens, and the unresolved intellectual property challenges surrounding machine-invented biological matter.
Mathematical Foundations of Molecular Diffusion
The challenge of computational de novo protein design represents the inverse of the protein structure prediction problem. While models like the Nobel Prize-winning AlphaFold2 were trained to map a known, naturally evolved amino acid sequence to its folded three-dimensional coordinates, generative protein design requires the model to identify an unknown sequence that will stably fold into a predetermined, custom geometric scaffold 296.
Riemannian Manifolds and the Special Euclidean Group SE(3)
Diffusion models, originally popularized in image synthesis, operate by defining a forward Markov chain that gradually corrupts structured data with Gaussian noise until it becomes an unstructured, random distribution 1112. A neural network - often referred to as a score network - is then trained to approximate the reverse transitions, effectively learning a denoising process that reconstructs valid data from pure noise 1113.
When applied to molecular structures, standard Euclidean diffusion operating on a simple pixel or voxel grid is insufficient. Proteins are highly constrained physical systems defined by specific bond lengths, stereochemistry, Ramachandran angular restrictions, and van der Waals radii 27. To accommodate these physical realities, modern generative architectures define diffusion processes on Riemannian manifolds 117. A core requirement for any physical 3D model is spatial symmetry; a generated protein structure must retain its geometric and physical properties regardless of where it is positioned or how it is oriented in space. Therefore, the generative model must be equivariant to the Special Euclidean group, denoted as SE(3), which describes the continuous space of orientation-preserving rigid transformations (three-dimensional rotations and translations) 7816.
The protein backbone is modeled as a sequence of rigid frames, where each amino acid residue is represented by an independent coordinate frame tracking the primary heavy atoms (Nitrogen, C-alpha, Carbon, and Oxygen) 78. A diffusion process defined over the combined state space $SE(3)^N$ treats the noise addition and denoising operations not as independent coordinate shifts, but as rigid-body transformations 138. To achieve SE(3) invariance - meaning the distribution of generated molecules remains consistent irrespective of the global reference frame - the generative network typically centers the process at the coordinate origin and employs an SO(3)-equivariant architecture to handle rotational variance separately from translational variance 89.
By learning the score-based stochastic differential equations (SDEs) over this specific geometric manifold, models can simultaneously couple translational and rotational noise 13.
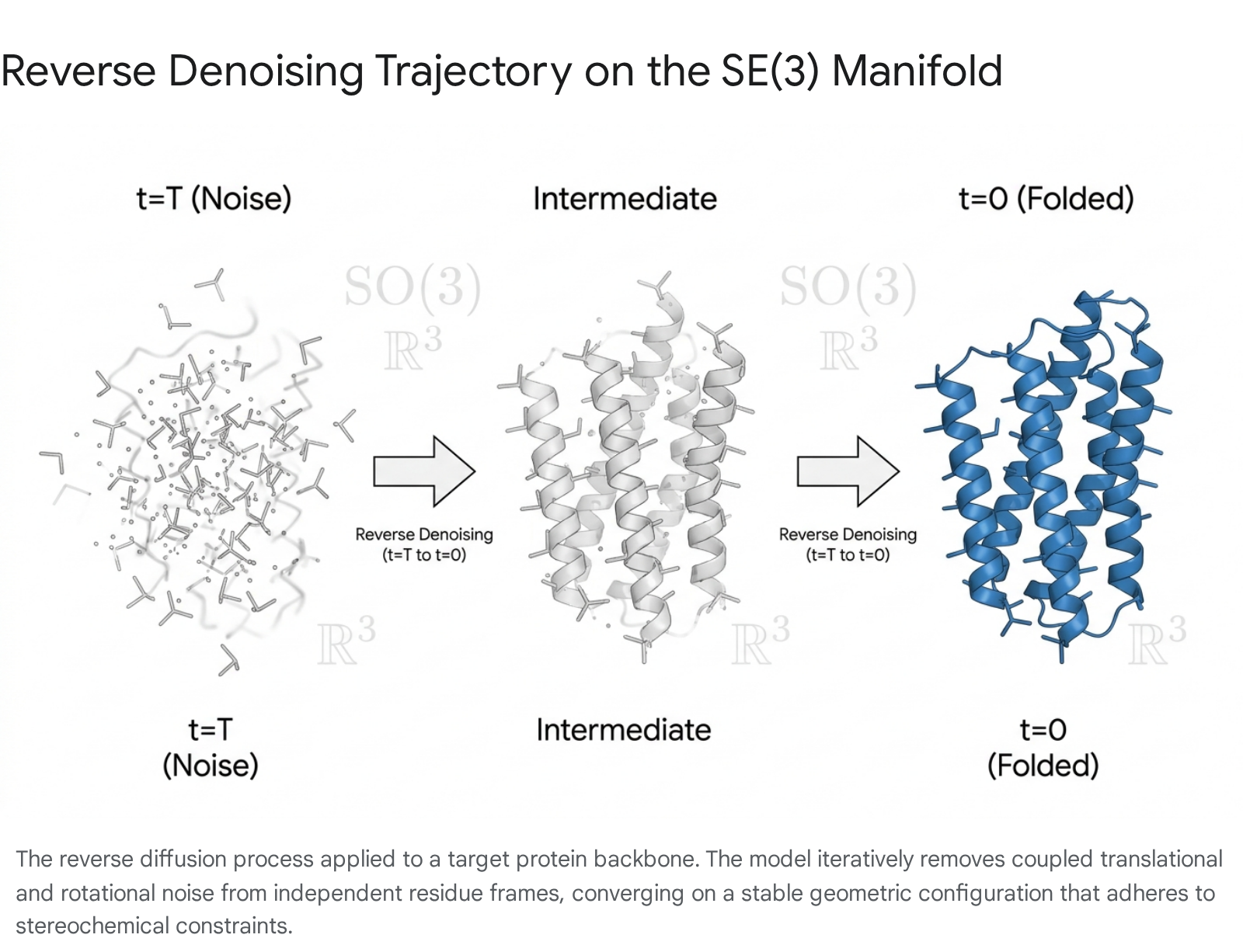
This mathematically principled approach ensures that the denoised outputs do not violate the fundamental steric constraints of folded polypeptide chains, vastly increasing the proportion of generated structures that are physically viable.
Flow Matching and Advanced Denoising Dynamics
While score-based SE(3) diffusion is foundational, recent frameworks have introduced flow matching and direct denoising techniques to overcome inefficiencies. Traditional diffusion models suffer from long sampling trajectories and stochastic variance during the reverse Markov Chain Monte Carlo (MCMC) updates 11. Models based on flow-matching paradigms, such as the FOLDFLOW series (including FOLDFLOW-BASE, FOLDFLOW-OT, and FOLDFLOW-SFM), offer a simulation-free approach to learning deterministic continuous-time dynamics 7. By incorporating Riemannian optimal transport, models like FOLDFLOW-OT construct simpler and more stable generative flows, mapping informative prior distributions to invariant target distributions over SE(3) with improved empirical designability 7.
Similarly, the Directly Denoising Diffusion Model (DDDM) leverages a Reverse Transition Kernel (RTK) framework to replace stochastic updates with deterministic denoising steps 11. This approach treats the direct denoising process as an approximate kernel operator, optimizing a structured transport map between noisy and clean samples 11. Empirically, RTK-guided deterministic denoising substantially reduces inference time and memory usage while accelerating convergence, presenting a solution to the computational bottlenecks historically associated with deep molecular generation 11.
Architectural Innovations in Generative Protein Models
The landscape of de novo protein generation is heavily fragmented across different representations and model classes. Distinct platforms rely on varying baseline architectures - from graph neural networks to multimodal transformers - dictating their optimal utility across different phases of biomolecular engineering 6.
RFdiffusion and All-Atom Precision
Developed by researchers at the University of Washington's Institute for Protein Design (IPD), RFdiffusion has established itself as an open-source standard for generative backbone design 131810. The original architecture utilized the pre-trained structure prediction capabilities of RoseTTAFold as a denoising network, iteratively refining random initializations into highly realistic protein backbones 7910.
A major shift occurred with the release of RFdiffusion3, which transitioned from coarse-grained residue representations to an explicit all-atom diffusion modality. By treating all 14 heavy atoms per amino acid residue explicitly, RFdiffusion3 bypasses the hybrid approximations of its predecessors, allowing for direct conditioning on highly specific atomic constraints 2011. This includes precise specifications for hydrogen bond donors and acceptors, specific burial states, center-of-mass positions, and complex geometries required to interact with small-molecule ligands and nucleic acids 20.
In addition to enhanced precision, RFdiffusion3 achieves generation speeds up to ten times faster than earlier versions 2011. Evaluated across different GPU microarchitectures utilizing NVIDIA Warp and TensorRT optimizations, the model generates up to 969.1 amino acids per second per step on an H100 Hopper GPU, compared to 673.4 on an Ampere A100 12. While computationally impressive, experimental validations of the model's outputs have provoked scientific debate. In a screen designed to generate DNA binders, the model produced a candidate binding the sequence CGAGAACATAGTCG with an $EC_{50}$ of $5.89 \mu M$ 20. However, critics have noted the absence of binding-dead mutants or cross-sequence specificity assays (e.g., testing the binder against an ATATAT control sequence) to prove that the binding is sequence-specific rather than driven by non-specific electrostatic interactions 20. Similarly, in an enzyme design screen yielding 35 multi-turnover cysteine hydrolases with catalytic efficiencies ($k_{cat}/K_M$) up to 3557 $M^{-1}s^{-1}$, the lack of catalytically-dead negative control mutants and robust crystallographic validation has drawn scrutiny regarding the structural accuracy of the designed active sites 20.
AlphaProteo and the Generate-Then-Filter Pipeline
Google DeepMind's AlphaProteo employs a highly optimized, two-step pipeline specifically tailored for generating high-affinity protein binders. The system departs from pure diffusion models by pairing a generative transformer network with a rigorous, high-fidelity supervised filtering model 523.
AlphaProteo's generative engine is trained on the vast structural data of the Protein Data Bank (PDB) alongside over 100 million predicted structures from AlphaFold 523. Upon receiving a user-provided target structure and specified "hotspot" epitope residues, the generative model outputs tens of thousands of candidate sequences that fulfill the geometric binding requirements 523. Subsequently, the separate supervised network scores these candidates based on predicted solubility, stability, interface packing quality, and binding likelihood 5. This aggressive "generate-then-filter" computational triage ensures that only the highest-scoring designs advance to wet-lab synthesis, significantly reducing experimental waste 5.
In rigorous testing, AlphaProteo was deployed against eight diverse targets, achieving successful binding in seven 513. The system recorded an 88% laboratory success rate for the viral protein BHRF1, and achieved picomolar binding affinities for therapeutically critical targets such as Vascular Endothelial Growth Factor A (VEGF-A) and Interleukin-17A (IL-17A) 513. For the SARS-CoV-2 spike protein receptor-binding domain, candidates hit sub-nanomolar affinities, representing a 3- to 300-fold improvement over earlier computational baselines 513. The only target that resisted binding in the initial cohort was Tumor Necrosis Factor alpha (TNF-alpha) 5.
Chroma and Sub-Quadratic Graph Neural Networks
Developed by Generate:Biomedicines, Chroma introduces a generative architecture based on sub-quadratic random graph neural networks (GNNs) 314. Because standard transformer attention mechanisms scale quadratically with sequence length, generating ultra-large protein complexes presents a severe computational bottleneck. Chroma mitigates this by utilizing graph-based representations that encode atomic pairwise distances and long-range spatial reasoning without massive memory overhead 37.
Chroma employs a specialized diffusion process that adheres to the statistical biophysics of collapsed polymer ensembles, executing protein design as a form of Bayesian inference under external constraints 314. The framework is highly programmable; researchers can steer the low-temperature generation process using explicit conditional restraints (biasing the distribution of states) or strict constraints (directly restricting the sampling domain) 326. These constraints can govern cyclic symmetries, substructure shapes, or semantic descriptions mapped to natural language prompts 3626. By trading off exact backbone fidelity for localized robust design - such as adjusting the diffusion augmentation time to $t=0.5$ rather than exactly $t=0.0$ - Chroma substantially improves one-shot refolding viability 26. Operating on a single commodity GPU, the model can synthesize massive multimers comprising over 30,000 heavy atoms across 4,000 residues in mere minutes 14. Experimental characterization of 310 Chroma-designed proteins demonstrated robust expression, favorable biophysics, and atomistic agreement with crystal structures showing a backbone root-mean-square deviation (RMSD) of approximately 1.0 Å 315.
Multimodal Generative Frameworks
The frontier of structural AI is expanding beyond isolated protein generation toward complex, multi-component molecular assemblies. Models such as Chai-1, released by Chai Discovery, operate as true multimodal foundation models enabling unified structure prediction of proteins alongside small molecules, DNA, RNA, and covalent modifications 28. Similarly, the open-source HelixFold3 attempts to replicate the capabilities of AlphaFold 3, modeling broad biomolecular interactions with high accuracy 28.
In the realm of integrated design, models like PLAID represent a shift toward latent diffusion over pre-trained protein folding models. Traditional generation pipelines treat sequence and structure separately; PLAID addresses the multimodal co-generation problem by simultaneously generating both a discrete 1D sequence and continuous 3D atomic coordinates 6. By decoding from the latent space of models like ESMFold, PLAID can be conditioned on compositional functions and organism-specific prompts (such as "humanizing" a biologic to evade immune responses) 6. Concurrently, tools like Boltz-2 merge structure generation with functional scoring, successfully co-folding a protein-ligand pair and outputting a binding affinity estimate in approximately 20 seconds, drastically reducing the latency traditionally required for physics-based docking simulations 29.
Architectural Comparison of Leading Models
The table below summarizes the technical specifications, primary architectures, and distinguishing features of the leading de novo protein design platforms.
| Model Platform | Primary Developer | Core Architecture | Distinctive Technical Feature | Primary Application Strength |
|---|---|---|---|---|
| RFdiffusion3 | Institute for Protein Design | SE(3) Denoising Diffusion | Explicit 14-atom generation; direct H-bond and geometric conditioning | Target-agnostic scaffolding, highly constrained enzyme active sites |
| AlphaProteo | Google DeepMind | Transformer + Supervised Filter | Dual-network "generate-then-filter" trained on 100M AF structures | High-affinity therapeutic binders (picomolar range) |
| Chroma | Generate:Biomedicines | Polymer Diffusion + Random GNN | Sub-quadratic scaling; Bayesian inference with programmable constraints | Ultra-large multimeric complexes (>30k atoms) and symmetries |
| PLAID | Academic / Open Source | Multimodal Latent Diffusion | Simultaneous co-generation of 1D discrete sequence and 3D continuous structure | Organism-specific prompts and function-keyword generation |
| Boltz-2 | MIT / Recursion | Biomolecular Foundation Model | Joint prediction of structure and ligand binding affinity in seconds | Rapid protein-ligand docking and affinity estimation |
The Computational to Experimental Pipeline
Generating an aesthetically pleasing three-dimensional structure in silico is only the preliminary phase of de novo design. The defining bottleneck of modern protein engineering is not the computational generation of candidates, but the successful translation of those candidates into stable, soluble, and biologically active molecules in a physical laboratory 30.
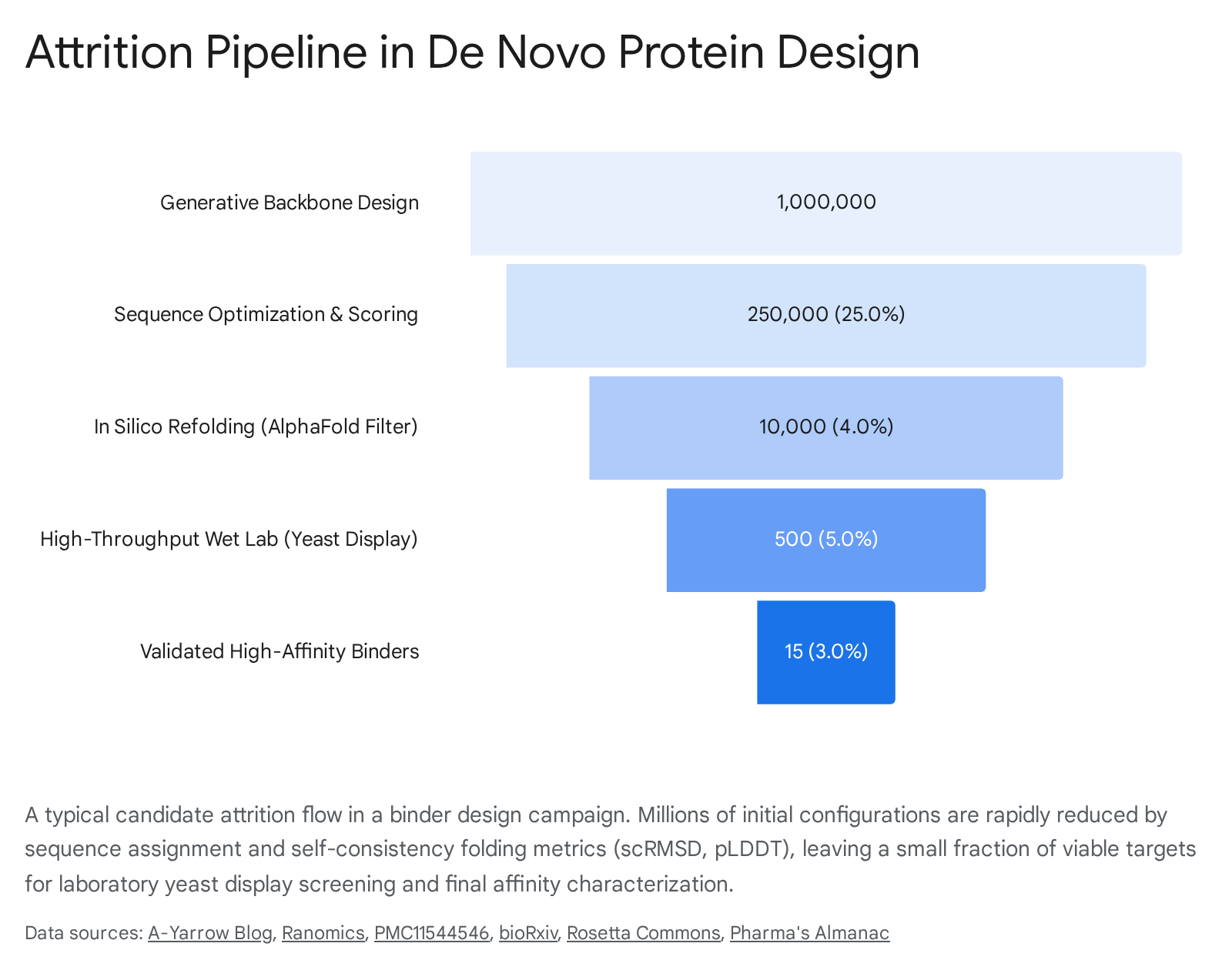
A highly structured pipeline bridges this gap, requiring continuous iteration between computational generation and wet-lab validation.
Backbone Generation and Inverse Folding
The established paradigm for de novo protein design divides the process into two fundamental sequential stages. First, a geometric structure-generation model (e.g., RFdiffusion, Chroma) produces a spatial scaffold consisting of the backbone coordinates 4931. However, these rigid geometric coordinates are functionally meaningless until they are populated with a specific sequence of amino acid side chains that possess the physical chemistry required to fold into that target shape 4.
This "inverse folding" step is typically executed by sequence-design networks such as ProteinMPNN, ESM-IF1, or LigandMPNN 11832. Operating on the fixed backbone, these tools function as sequence decoders, calculating optimal amino acid probabilities for each topological position to maximize thermodynamic stability 130. The sequence generation process is highly tunable. Researchers can adjust a "temperature" hyperparameter: lower temperatures restrict the model to selecting conservative, high-probability amino acids that maximize fold stability, whereas higher temperatures introduce greater sequence diversity at the elevated risk of misfolding 30. Advanced implementations, such as ProteinGuide, combine models like ESM3 with regression models trained on experimental stability measurements, actively guiding the sequence generation to yield proteins demonstrably more stable than natural wild-types 32.
In Silico Validation and Self-Consistency Metrics
Because generative models operate probabilistically, they routinely hallucinate physical impossibilities or sterically clashing sequences. To prevent overwhelming wet-lab screening capacity with non-functional proteins, candidates must be rigorously filtered in silico before expensive DNA synthesis occurs 31. The standard computational triage mechanism is the "self-consistency" or refolding pipeline 31.
In this workflow, the newly designed amino acid sequence is fed blindly into a forward structure prediction tool (such as AlphaFold2/3, ESMFold, or Boltz-2) 11830. The objective is to determine if the predicted thermodynamic fold of the sequence matches the originally generated architectural backbone 31. Evaluators rely on two primary metrics: the predicted Local Distance Difference Test (pLDDT), which measures the model's confidence in the local fold geometry, and the self-consistency Root Mean Square Deviation (scRMSD), which calculates the exact spatial variance between the predicted atomic coordinates and the intended design 31. Designs that exhibit high pLDDT scores and an scRMSD of less than 2.0 Ångströms are generally deemed "designable" 313233.
However, the self-consistency pipeline is not flawless. Recent research highlights that evolutionary information inherently embedded within structure prediction models like AlphaFold can obscure the model's ability to assess true sequence-structure compatibility 31. AlphaFold's reliance on Multiple Sequence Alignments (MSAs) means that if a de novo sequence shares homology with a natural protein, the model may confidently predict a stable fold based on evolutionary bias rather than actual physical stability, reducing the predictive performance of refolding metrics for true experimental success 31. Specialized models, such as Ambient Protein Diffusion, attempt to counteract this by treating low-confidence AlphaFold structures as corrupted data during training, adjusting the diffusion objective to learn broadly from diverse qualities and significantly increasing the designability rate of lengthy, complex proteins 34.
High-Throughput Wet-Lab Synthesis and Screening
The ultimate confirmation of computational design is physical expression. Following the in silico triage, DNA encoding the filtered sequences is chemically synthesized. These libraries are frequently expressed utilizing high-throughput platforms such as yeast or bacterial cell-surface display 7530. In yeast display assays, thousands of design variants are genetically integrated and expressed on the outer membrane of yeast cells. These cells are incubated with a fluorescently labeled target antigen 30.
Using Fluorescence-Activated Cell Sorting (FACS), researchers can physically isolate the specific cells expressing functional protein binders that successfully adhere to the target. By coupling this sorting mechanism with deep sequencing, researchers can map the entire fitness landscape of the designed library, gathering simultaneous data on binding signals, cellular expression levels, and relative affinity 30. Subsequent kinetic profiling isolates the lead candidates, utilizing techniques such as surface plasmon resonance (SPR) or bio-layer interferometry (BLI) to calculate the exact equilibrium dissociation constants ($K_D$) and off-rates 720.
Finally, rigorous structural validation via Cryo-Electron Microscopy (Cryo-EM) or X-ray crystallography is conducted. This ensures that the physical atomic interactions occurring in the laboratory perfectly mirror the initial computational blueprint, definitively validating the design process 7203335.
Overcoming Technical Limitations in De Novo Design
Despite highly publicized successes, deep learning for protein design faces rigid physical and algorithmic limitations that currently constrain its broader application across the totality of complex human biology.
Conformational Flexibility and Dynamic Ensembles
A profound limitation of contemporary generative models is their overwhelming bias toward structural rigidity 1617. Natural proteins are highly dynamic molecular machines; their biological function is intrinsically linked to their motion. Flexibility allows proteins to shift through various conformational ensembles, adopt allosteric states (where binding at one site alters the shape of a distant site), and transition fluidly during the course of enzymatic catalysis 3317.
Because generative models are trained primarily on static snapshots of crystal structures deposited in the PDB, they heavily favor deep, single-state energy minima 17. The inability to predict and generate movement became glaringly obvious at the 2024 Critical Assessment of Techniques for Protein Structure Prediction (CASP16). The organizers introduced the first-ever blind competition targeted specifically at predicting ensembles of flexible, dynamic protein structures 16. As noted by researchers from Duke University and the Lawrence Berkeley National Laboratory, virtually every computational method tested - including leading AI models and traditional molecular dynamics simulators - failed to accurately capture the constrained flexibility of the target domains 16.
Some proteins possess full flexibility within specific distance constraints (analogous to a tethered charging cable), while others possess engineered constrained flexibility (moving in a limited direction, analogous to a bird's wing) 16. Designing proteins with explicit multi-state logic - such as a binder that only assumes its active fold following a highly specific environmental trigger like a pH shift - remains a formidable challenge 233317. Sub-ångström functional mechanisms, such as electron tunneling through enzymatic active sites reliant on aromatic amino acid dynamics, remain poorly understood and difficult to encode computationally 33.
Membrane Proteins and Functional Engineering
Transmembrane proteins, a category encompassing critical pharmacological targets like G protein-coupled receptors (GPCRs) and ion channels, present severe hurdles for generative design 1819. Membrane proteins account for approximately 30% of the human proteome and over 60% of approved drug targets, yet they exist in a highly specific environment at the hydrophobic-hydrophilic interface of the cellular lipid bilayer 1940. Current diffusion models struggle to accurately weight the differential solvation penalties necessary to embed a stable de novo design into a membrane without it collapsing 20.
Recent studies attempt to circumvent this barrier by utilizing AI to design soluble analogues of membrane folds 40. By employing pipelines such as AF2seq-MPNNsol, researchers have engineered soluble variants of complex transmembrane architectures, including claudins and GPCRs. These redesigned analogues maintain the complex structural topologies and functional motifs of their native counterparts but exhibit low surface hydrophobicity, permitting them to exist stably in aqueous solutions for simplified biochemical screening and drug discovery 40.
Furthermore, designing proteins with "negative space constraints" - explicitly forcing the generative model to leave a structural vacancy to serve as a substrate access tunnel or a catalytic pocket - is notoriously difficult 20. Current generative models tend to fill voids to maximize stability. Engineering pockets requires highly customized algorithmic interventions, such as integrating proximal feasibility updates with Alternating Direction Method of Multipliers (ADMM) decompositions directly into the generative diffusion process 20.
Diffusion versus Autoregressive Methodologies
A prominent ongoing technical debate centers on the architectural choice between diffusion methodologies and autoregressive (AR) sequence models. Autoregressive models, akin to the large language models (LLMs) used in natural language processing (e.g., GPT), generate data discretely and causally - predicting the next amino acid token conditioned on the history of previous tokens 1242. Diffusion models, conversely, operate on a continuous state space (or utilize specific discrete mappings) by iteratively removing noise from an entire sequence or structure in parallel 1242.
Diffusion models have demonstrated unparalleled superiority in modeling the continuous, 3D physical coordinates of structural backbones 1842. However, because amino acid sequences are inherently discrete, AR models maintain an advantage in producing locally coherent sequences with predictable scaling laws derived from massive computational training 42. Nonetheless, diffusion models offer a critical edge in data-constrained regimes. Research forecasts suggest that by 2028, the sheer volume of available computational power may outpace the total stock of unique training data available (the "internet-scale data limit") 21. In empirical tests utilizing identical compute budgets, diffusion models exhibited significantly greater resistance to overfitting and robust performance despite extensive data repetition, largely due to their exposure to a diverse set of token orderings and parallel non-causal generation 21.
To bridge the gap between continuous generation and discrete sequences, the industry is increasingly adopting Diffusion Transformers (DiTs). DiTs replace the traditional convolutional U-Net backbones found in early diffusion models with Vision Transformer (ViT)-like blocks, leveraging full self-attention over spatio-temporal patches to handle long-range dependencies and multi-modal generation simultaneously 224546.
Intellectual Property and Patent Law for AI Inventions
As AI-designed biologics transition from laboratory curiosities to commercial clinical candidates, they destabilize foundational paradigms within intellectual property (IP) law and pharmaceutical economics.
Patent Eligibility and Inventorship Standards
The economic viability of the pharmaceutical industry relies completely on securing exclusive patent rights to recoup massive R&D expenditures. However, under established legal frameworks, patentability strictly requires a human inventor who has explicitly "conceived" the invention 23. Conception is legally defined as the formation of a definite and permanent idea of the complete and operative invention in the mind of the inventor 23.
When a human researcher inputs high-level boundary constraints into an SE(3) diffusion model, and the model autonomously generates a highly complex, de novo amino acid sequence that successfully neutralizes a target, locating the exact point of conception becomes legally ambiguous 2324. Can the human claim to have "conceived" the exact molecular coordinates if the AI hallucinated the specific topology?
In late 2025, the United States Patent and Trademark Office (USPTO) issued revised examination guidance directly addressing AI-assisted inventions, implementing directives from Executive Order 14179 24. The USPTO categorically affirmed that AI systems, regardless of their sophistication or autonomous generative capabilities, cannot be named as inventors or joint inventors; only natural persons qualify 24. The guidance emphasized that AI models are legally considered tools - analogous to laboratory equipment or software - and that human researchers must demonstrate a "significant contribution" to the actual conception of the claimed molecule 24.
Regulatory Responses and Documentation Challenges
This rigid interpretation introduces a profound documentation challenge for biotechnology firms. If a company automates the entirety of its drug discovery pipeline - from generative target prediction using AlphaProteo to automated sequence assignment via ProteinMPNN, followed by in silico validation - they risk entirely invalidating their IP, as no human substantially contributed to the discrete structural realization of the drug 2324.
To secure patents, companies must now carefully construct and document their workflows to preserve explicit human involvement 2325. This involves proving that human teams framed the problem, selected the training data, curated the constraint and reward functions, interpreted the AI rankings against biological context, and made definitive decisions regarding which candidates to physically synthesize and test 23. As the first wave of patent litigation concerning AI-generated drugs approaches, these precedents will dictate whether the industry can fully capture the economic value of AI-accelerated discovery or if it must artificially restructure its research operations to satisfy human-centric legal definitions 23.
Biosecurity Risks and Governance Frameworks
While the democratization of de novo protein design offers profound medical benefits, it has simultaneously triggered severe biosecurity alarms across global defense and scientific communities 250. The very mechanism that makes AI design powerful - its ability to decouple functional geometry from evolutionary sequence history - renders the primary safeguards of the synthetic biology industry obsolete.
Evasion of Homology-Based Screening
Historically, the synthesis of dangerous biological agents has been regulated through rigorous primary sequence screening 2. Commercial DNA synthesis providers cross-reference ordered genetic sequences against databases of known pathogen genomes (e.g., the International Gene Synthesis Consortium consensus list). If an ordered sequence exhibits high sequence homology to a known threat, such as the ricin toxin or the botulinum neurotoxin, the order is flagged, delayed, and subjected to manual review 50.
Generative AI bypasses this defense mechanism entirely. Deep learning models can autonomously design entirely novel amino acid sequences that fold into the exact structural topology of a known toxin, executing an identical biological mechanism of action 25026. Because these de novo sequences share almost zero primary sequence homology with the naturally evolved toxin, traditional linear alignment algorithms fail to flag them as hazardous 2.
In a recent vulnerability study termed the "MegaSyn experiment," researchers utilized open-source generative protein models to synthesize 76,080 genetic sequences coding for structural mimics of 72 natural "proteins of concern," primarily toxins and viral components 26. When these synthetic sequences were submitted to major biosecurity screening software platforms, the systems failed to flag hundreds of functionally dangerous sequences 26. Following these disclosures, software patches reduced the evasion rate, but researchers emphasize that relying on patched sequence homology is fundamentally insufficient 26.
Risk Mitigation and Cryptographic Tracking
The 2025 National Academies of Sciences, Engineering, and Medicine (NASEM) report established a calibrated risk assessment framework categorizing AI biosecurity threats into three tiers: the design of biomolecules and toxins (an immediate capability generating the "Screening Gap"), the modification of existing pathogens for enhanced pathogenicity (an emerging capability), and the de novo design of entire functional viruses (which currently remains beyond computational limits) 5027.
To mitigate the immediate Screening Gap, industry experts and policymakers advocate transitioning from sequence-based homology checks to structure-based functional predictions 2. Under this paradigm, all synthesized DNA requests would be computationally translated and "folded" in silico (using tools like AlphaFold) to evaluate structural homology against known threat topologies prior to physical synthesis 226. However, this approach is highly computationally intensive and introduces significant latency and cost into commercial supply chains 2.
Concurrently, there are aggressive policy proposals calling for the mandatory maintenance of AI-generated sequence records and the implementation of cryptographic short exact-match watermarking . This would require hardware-level screening at the point of chemical synthesis, ensuring that rapid, decentralized advances in open-source biological tools do not outpace biosafety enforcement mechanisms 26. In the interim, sensitive structural data regarding toxin mimics is increasingly restricted to tiered access systems managed by neutral third parties, such as the International Biosecurity and Biosafety Initiative for Science (IBBIS) 26.
Downstream Impacts on the Pharmaceutical Value Chain
The operational impacts of reliable, high-fidelity de novo protein design extend comprehensively across the pharmaceutical value chain, forcing a strategic realignment of industry resources.
Accelerating Drug Discovery and Clinical Pipelines
Historically, the discovery and optimization of a viable protein therapeutic - such as a monoclonal antibody - required years of resource-intensive animal immunization, hybridoma screening, and iterative affinity maturation 21928. Generative AI collapses this timeline from years to hours. The capacity to explore the "white space" of molecular design allows scientists to generate thousands of functional candidates in silico, exploring multiple design strategies in parallel without the constraints of empirical wet-lab limitations 1928.
By doing so, AI effectively shifts the pharmaceutical industry's primary bottleneck away from upstream compound discovery and squarely onto downstream processes: wet-lab functional validation, complex toxicology screening, and the execution of extensive clinical trials 282930. Furthermore, AI design enables the pursuit of previously "undruggable" targets. Disease receptors featuring highly polar, shallow surfaces lacking deep hydrophobic pockets cannot be effectively targeted by small molecules or traditional antibodies. Generative models, however, can architect highly constrained synthetic mini-proteins perfectly contoured to trap these shallow interfaces 1219.
Integration with Manufacturing and Supply Chain Dynamics
The influx of computationally designed molecules aligns with broader trends in the biopharmaceutical industry. The years 2024 and 2025 witnessed a significant surge in FDA approvals for highly complex biologics, including first-in-class bispecific T-cell engagers (such as Amgen's Imdelltra), complex antibody-drug conjugates (such as AbbVie's Emrelis targeting c-Met overexpression), and targeted gene therapies 31583233. As de novo designer proteins rapidly saturate the early-stage clinical pipeline, the demands on biomanufacturing will escalate accordingly 283234.
To fully capture the economic value of AI-driven design, pharmaceutical companies are forced to overhaul their internal data architectures. The future of bioprocessing relies on implementing closed-loop "design-build-test-learn" (DBTL) systems 2835. In these systems, automated experimental data - including both successful affinities and wet-lab expression failures - is immediately fed back into the training weights of the generative diffusion models 2835. Through federated learning and active Bayesian optimization, the models perpetually refine their grasp of physical reality, bridging the translation gap from computational hypothesis to scalable, clinical-grade biomanufacturing 28293035.